
Three months ago, FDA committed to fully eliminate a backlog of about 200 orphan drug designation requests that were pending review with FDA, and to implement policies that would require FDA to respond to all new designation requests within 90 days of receiving them. Moreover, the agency pledged to never allow a backlog of these designations to accumulate again.
At that same time, I said that we may pursue other policies that we believe would enable us to better advance the goals of the Orphan Drug Act (ODA). All of these commitments were part of a new Orphan Drug Modernization Plan that we announced on June 29th.
I’m pleased to update you on our progress in meeting each of these objectives.
First, owing to the dedicated efforts of the orphan drug designation team who oversee the Orphan Drug Designation Program, the first of these goals has been fully achieved. Reviews of all orphan drug designation requests older than 120 days were completed on August 28th. This was well ahead of the September 21st deadline that we had set for ourselves under our orphan drug plan. The achievement is hopeful news for those with a rare disease, defined as a disease which generally affects fewer than 200,000 people in the United States. Companies that receive orphan drug designation for their product qualify for various incentives including tax credits for clinical trial costs, relief from prescription drug user fees and the potential for seven years of marketing exclusivity after the drug is approved.
Second, we’re putting in place new policies to improve the efficiency of our review process to ensure that we meet our new 90-day mandate to prevent new backlogs.
To do this, FDA will pursue a range of process improvements. Among them: we’ll reorganize our review staff to improve workload efficiencies and to better leverage the expertise across FDA’s medical product centers. We’ll also use lean management principles to design a new process map that’s based on an assessment of sources of delay or redundancy and metrics for measuring success. This new workflow will outline a more efficient process that eliminates redundancies and delays that don’t add value. We’ll share this map with you in the fall.
Finally, we’re going to be taking new policy steps to make sure that the incentives offered by the ODA are granted by FDA in a way that’s consistent with the manner Congress intended. To that end, FDA will soon hold a public meeting to get input on complex scientific and regulatory issues such as those raised by molecularly targeted drugs and biologics and the appropriate application of orphan incentives in that paradigm. As part of this process, FDA will also be examining aspects of how the agency grants designations, to make sure they continue to reflect current science and drug development and the goals intended by Congress.
For all the success of the ODA, there’s been criticism that some sponsors are using designations as a way to sidestep other important public health goals set out by Congress. We need to make sure our policies take notice of all of these new challenges and opportunities.
FDA plans to advance certain guidance documents and other policies to address these issues. One guidance document will close a loophole that allows sponsors to avoid an obligation to study drugs in pediatric indications. This circumstance arises if sponsors received an orphan designation for a pediatric subtype of an otherwise common and non-orphaned adult disease.
The longstanding practice of allowing pediatric subpopulations of common diseases to be designated as orphan conditions was intended to promote pediatric drug development. It pre-dated the implementation of the Best Pharmaceuticals for Children Act and the Pediatric Research Equity Act (PREA), two laws that were specifically aimed at promoting more pediatric studies.
Now, instead of instigating more pediatric research, the granting of the orphan designation in the pediatric subpopulation can have the opposite effect. It can allow sponsors to sidestep pediatric study requirements that are part of other laws aimed at promoting this same research.
Consider a condition like inflammatory bowel disease. A drug may be approved to treat the large population of adults with the condition. Then the same drug may be granted an orphan designation to treat the much smaller population of a subset of children suffering from the same condition.
But once a drug receives an orphan designation for a pediatric population of the adult disease, the drug then becomes statutorily exempt from the requirements of PREA. This occurs even in cases where the sponsor never goes on to develop the drug for this pediatric use. These PREA requirements could have required the sponsor to study the drug for this or other uses in children. By granting the drug a pediatric orphan designation, it means the drug never has to actually be studied for a pediatric use. It’s a loophole that is in direct opposition to what Congress intended.
Nobody envisioned this unintended conflict between the original ODA and the provisions outlined in PREA. In effect, by letting sponsors designate pediatric subpopulations of drugs intended to treat adult diseases, the drug makers receive an unintended “free pass” from having to study drugs in these or other pediatric uses. Thus, rather than ensuring more pediatric research, as Congress envisioned, we can end up with fewer pediatric studies. FDA will soon issue a draft guidance document that’s aimed at closing this inadvertent loophole.
Taking these and other steps to modernize our stewardship of the ODA is imperative. Science is giving us new opportunities to use the tools offered by the ODA to advance innovation in more areas of medicine where patients have few, if any options. At the same time, the demands on FDA’s orphan drug program continue to grow. We need to make sure the policies governing how we implement these key provisions are modern and efficient.
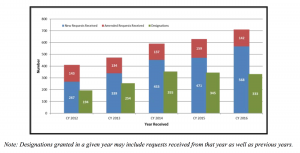
The number of requests received by FDA under the Orphan Drug Designation Program has steadily increased over the past five years, rising to 568 new requests in 2016. This is more than double the number of requests received in 2012. Patients with rare disease often have limited or no treatment options. We want to maximize new opportunities for patients.
FDA will continue to make full use of tools provided by Congress to apply incentives for the efficient development of rare disease therapies, and for activating more pediatric research. These steps will help us achieve our ultimate goal: to facilitate the development of safe, effective innovations that have the potential to meaningfully impact rare diseases.
Scott Gottlieb, M.D., is Commissioner of the U.S. Food and Drug Administration