
Small biopharmaceutical companies are becoming increasingly important as drivers of innovation in drug development. It has recently been estimated that the majority of drugs currently in development are in the hands of small biopharmaceutical companies.1 Such companies range in size from virtual companies with no commercial products and no revenue to those with only a few commercial programs.
Small biopharmaceutical companies often encounter important challenges in designing and implementing clinical development programs. In a context in which only approximately 10% of clinical programs result in drugs that achieve regulatory approval, small-company clinical programs may have an even lower rate of success than that of large companies2 owing to limited internal experience in clinical development and limited infrastructure, which may also affect manufacturing and clinical supply. However, these challenges are largely overshadowed by limited resources and funding, which in turn fuel demand for short timelines owing to the need to demonstrate progress to investors. As such, these companies must focus their resources on small, less-costly development programs for very specific targets and often must spearhead new approaches to testing new products in order to survive.
Small companies use a variety of approaches to address these challenges, including the use of new technical platforms, the use of new formulations or technologies that enhance the actions of known drugs, or the use of trial designs that take advantage of the specific market they hope to enter. Other companies develop products that are spun off from or licensed from large companies. In fact, many small companies may choose to partner with larger companies to add resources and experience.
Many small companies also repurpose drugs or pursue narrow niche markets — such as rare inherited diseases, uncommon cancers, or specific infectious diseases — to remain viable. Furthermore, many companies turn to rare diseases for an opportunity to successfully negotiate many of the aforementioned issues, though such diseases present their own challenges — in particular, the small number of patients available for clinical trials. Table 1 illustrates some examples of approaches used by small companies that achieved Food and Drug Administration (FDA) approval for their drugs in 2014 and 2015.
It is beyond the scope of this article to provide a comprehensive review of all the challenges and strategies used by small biopharmaceutical companies. Herein we provide an in-depth discussion of several examples, with an emphasis on drugs developed for rare diseases.
Use of a Historical Control Group When the Use of Placebo Is Problematic
Although randomized, double-blind, placebo-controlled clinical trials are the standard in clinical development and usually present the quickest way to discern a treatment effect, severe and uniformly rapidly progressive, life-threatening diseases may present a challenge to the acceptability of placebo controls when preliminary data suggest a lifesaving potential. Pompe disease results from a deficiency of the enzyme acid α-glucosidase (GAA). GAA degrades lysosomal glycogen, and if there is insufficient GAA activity, glycogen accumulates in cardiac and skeletal muscle, causing progressive cardiomyopathy and generalized muscle weakness and hypotonia, which result in severely delayed motor development and cardiorespiratory failure.3 Patients may begin to exhibit signs and symptoms in the first few days of life or as late as the sixth decade. The most severe and rapidly progressive form is infantile-onset Pompe disease, in which clinically significant manifestations generally develop within the first months of life. If the condition is left untreated, 70 to 80% of children with infantile-onset Pompe disease die from cardiac or respiratory failure before 1 year of age.3
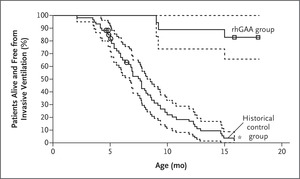
Figure 1. Kaplan–Meier Analyses of Survival Free from Invasive Ventilation (Primary Efficacy End Point).
Enzyme-replacement therapy with recombinant human GAA (rhGAA, Myozyme) was approved for the treatment of Pompe disease in 2006. This approval was based on a study that evaluated the effects of rhGAA in 18 infants with severe infantile-onset Pompe disease; these patients exhibited cardiomyopathy and profound deficiency of GAA activity at an age of 7 months or younger. It was considered unethical to include a placebo group as part of the study design, because infantile-onset Pompe disease is a rapidly fatal disorder and early clinical trials had shown that treatment with various forms of rhGAA can improve survival, cardiac and respiratory function, and growth and motor development in severely affected infants.4 Instead, this study used a historical control group as a comparator. The authors identified a historical control group of 61 severely affected infants 6 months of age or younger by applying the study inclusion and exclusion criteria to a group of 168 patients with infantile-onset Pompe disease who were identified through a retrospective chart view. The cohort included 168 patients from nine countries and 33 different sites who were born during a time period in which contemporaneous care was available that was similar to the care available for the 18 infants in the study group. Overall survival and survival free from invasive ventilation were compared between treated patients and this historical cohort; the sample size of 18 treated patients provided more than 95% power to detect an absolute difference of 60 percentage points in overall survival between treated and control groups. Patients who received rhGAA treatment for 52 weeks had a markedly higher survival rate than the historical control group, and they had better respiratory function and less evidence of cardiomyopathy; a subgroup of patients also had better motor function (Figure 1).5
The use of a historical control group was acceptable for regulatory purposes owing to the objective and well-defined end point and the large treatment effect observed. Enzyme-replacement therapies for lysosomal storage disorders may result in such large effects on disease, which justifies the use of such an approach. Admittedly, many drugs in development do not produce such dramatic treatment effects.
Use of a New Biomarker End Point That Is Likely to Predict Clinical Benefit
A frequent challenge for small companies pursuing orphan niche products is that many of these disorders lack previously recognized end points and have poorly characterized, heterogeneous rates of disease progression. Without such knowledge, it is difficult to determine the sample size and duration of a clinical trial.
Fabry’s disease is another lysosomal storage disorder, but it is caused by an X-linked mutation in the gene encoding α-galactosidase. In male patients, the disease has a pronounced vascular phenotype with effects most commonly in the kidney, heart, and brain, owing primarily to storage of globotriaosylceramide (GL-3) within the endothelium; the disease results in renal failure, cardiomyopathy, cardiac events, and strokes. Prolonged and heterogeneous rates of disease progression among patients with Fabry’s disease complicate the design of clinical trials and typically necessitate large cohorts that are followed for very long time periods to discern between-group differences in clinical outcome end points. A new surrogate end point could allow greater homogeneity among patients in baseline assessments and response to therapy, with a smaller sample and shorter duration of study.6
GL-3 storage in the endothelium is directly related to the cause of eventual renal failure that affects most patients with Fabry’s disease. In animal models and a phase 1 study, analysis of GL-3 granules that were present in glomerular endothelial cells in renal biopsies showed that a recombinant enzyme could clear GL-3 storage and return the endothelial cells to near-normal, if not normal, status in a dose-dependent manner within a few months.6
The challenge of using GL-3 clearance in glomerular endothelium as a surrogate end point was that biopsy data can be quite variable when samples are obtained from different areas of the kidney and the scoring can be subjective. Extensive work on multiple biopsies and scoring systems, adjudication of results, and meetings with FDA reviewers were needed to gain agreement on the reliability of biopsy and on the extent of GL-3 clearance that could be considered to be clinically meaningful to support this pathological assessment as a surrogate end point.
It was reasoned that a reduction to normal or near-normal amounts (scored as 0 on a 4-point scale) would be likely to predict clinical benefit.7 The investigators chose this end point and used a panel of three renal pathologists to score the biopsy material. The pathologists independently assessed slides of renal biopsies that were presented in a blinded and randomized fashion. All the pathologists participated in preliminary sessions to establish the criteria for the grading scale that was used to evaluate clearance from affected tissues.8The results of this trial showed a significant treatment effect: 20 of the 29 treated patients had renal biopsies with a score of 0, whereas none of the 29 patients who received placebo did (P<0.001). Similar results were observed in the endothelium in heart and skin biopsy samples.
The FDA accepted the argument that the reduction to normal or near normal in GL-3 accumulation in renal glomerular endothelial cells is reasonably likely to predict clinical benefit, and the product received accelerated approval after the results of this randomized, placebo-controlled study using biopsy results as an end point were disclosed.7
Predictive Enrichment with the Use of a Genetic Marker
Today “targeted therapies” offer more precise ways to identify patients who are likely to benefit from treatment. In the past, therapies with promise were studied in a large population with the hope that enough participants would have a response to make the trial successful, and a broad indication for use would be granted. However, if a therapy benefited only a small number of patients, such trials might fail.
Cystic fibrosis is a progressive lung disease that provides a context to illustrate clinical-trial design that takes advantage of a targeted patient population. The disease is caused by mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) protein, an epithelial ion channel that is involved in salt and fluid transport in multiple organs, including the lung.9 Some mutations permit the CFTR protein to reach the epithelial-cell surface, but the protein is defective in chloride transport. The most prevalent mutation of this type in patients with cystic fibrosis causes a substitution of glycine for aspartic acid at amino acid 551 (G551D-CFTR mutation); this mutation occurs in only 4 to 5% of persons with cystic fibrosis.10
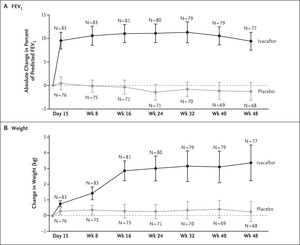
Figure 2. Changes in FEV1 and Weight with Ivacaftor.
Pharmaceutical agents that increase the ion-channel function of activated cell-surface CFTR are referred to as “potentiators.” Ivacaftor is an approved oral CFTR potentiator and was tested in a randomized, double-blind, placebo-controlled trial. In this trial, 167 patients who tested positive for the targeted mutation were randomly assigned to either 150 mg of ivacaftor or placebo every 12 hours. The change from baseline through week 24 in the percent of predicted forced expiratory volume in 1 second (FEV1) was greater by 10.6 percentage points in the ivacaftor group than in the placebo group (P<0.001), and a significant treatment effect was maintained through week 48 (Figure 2).10,11 Ivacaftor was also associated with significant benefits with respect to average weight gain, concentration of sweat chloride (a measure of CFTR activity), and freedom from pulmonary exacerbations. This study suggests that a drug targeting CFTR dysfunction can affect lung function and symptoms and thus confirms that CFTR is a valid therapeutic target. This is a great example of predictive enrichment — that is, the study used genetics to identify a population of patients with cystic fibrosis who were likely to have a response to this treatment.
As suggested above, identifying the patients who are likely to have a response to treatment, and then studying them, greatly enhances the power of a study and has clear implications for how a drug will be used. The use of such patient-selection tools can greatly reduce the number of patients needed to show efficacy. It can be especially critical when those who are likely to have a response are only a small fraction of all patients with a disease (e.g., 4 to 5%). In such a case, finding an effect in an unselected patient population may be practically impossible.
A High Placebo Response Rate and the Importance of Controlled Trials
Up to this point, we have focused on successful strategies and orphan diseases. A common error of many small companies is an overemphasis on the results of early, uncontrolled trials for planning and business decisions. To illustrate this error, we turn to an example from Parkinson’s disease.
Transplanted human fetal mesencephalic tissue survives and has antiparkinsonian effects in some patients with advanced Parkinson’s disease.12 However, practical and ethical issues preclude widespread use of fetal tissue as a therapeutic option for Parkinson’s disease. So, before initiating a large-scale confirmatory trial, a small biotechnology company (Diacrin, Charlestown, MA) conducted an uncontrolled trial involving 12 patients with Parkinson’s disease.13 These patients received unilateral implants of embryonic porcine mesencephalic tissue into the caudate and putamen. Half received cyclosporine treatment for immunosuppression, and half received fetal tissue treated with a monoclonal antibody directed against major histocompatibility class I.
At 1 year, there was significant decrease in the total score on the Unified Parkinson’s Disease Rating Scale (UPDRS; scores range from 0 to 199, with higher scores indicating greater disease severity) in the “off” state (i.e., while medication effect is at its least), with an adjusted mean decrease of 19% from baseline (mean score, 83.7 at baseline vs. 66.8 at 1 year). The “off” score on the UPDRS subscale for activities of daily living (range, 0 to 52, with higher scores indicating greater disease severity) also decreased, from a mean of 27.1 before surgery to a mean of 20.4 at 1 year after surgery. Only two patients, both in the cyclosporine group, individually showed large decreases, but 18F-fluorodopa positron-emission tomography failed to show changes on the implanted side of these two patients. In two patients who died, small numbers of implanted cells that included dopaminergic neurons and other porcine neural and glial cells were detected at postmortem analysis.
Although changes in UPDRS motor scores were considered to be “objective” results, the study was very small, and the sponsor was urged to conduct a multicenter, prospective, randomized, double-blind, placebo-controlled study to evaluate this therapy further. The FDA suggested that the use of sham-surgery controls — in which burr holes are created without further experimental intervention — is the preferred method to test new neurosurgical inventions for Parkinson’s disease. The primary end point of the confirmatory trial was the difference between the treatment group and the control group in the change in the total UPDRS score in the “off” state, 18 months after surgery. The 10 treated patients showed a mean decrease of 24.6±25.0%, but the 8 control patients surprisingly showed a mean decrease of 21.6±14.0% (P=0.60). No significant between-group differences were observed on four secondary end points.14
One of the possible reasons for the disappointing results of this trial might be the high placebo response rate (overall rate, 16%; range, 0 to 55) that has been documented in several surgical trials in Parkinson’s disease.15 To account for a placebo effect, a sufficient number of patients and a defined patient population are needed. Because of the limited resources that were available to a small biotechnology company such as Diacrin, it had to take major risks to achieve its stated goal (i.e., expecting a large treatment effect with a small number of patients). The goal can be met only if there is a large treatment effect with little or no placebo effect. Soon after the announcement of the trial results, Diacrin could not remain a viable company and closed the further development of this therapy in patients with Parkinson’s disease. Unfortunately, this is a not uncommon outcome for many small companies.
Conclusions
The unique challenges of clinical development by small companies are often addressed with the use of smaller clinical development programs than those used by large companies. Development of drugs for rare diseases may be used as a strategy, but the small size of the populations with such diseases and the small samples available for trials require approaches that can maximize the power to detect efficacy, which can include the use of historical controls, new surrogate end points, or enrichment for participants who are likely to have a response. Temptations to use uncontrolled, early, small studies to support further development of products may prove problematic. Small companies with limited resources require both innovative approaches and rigor for success.