What’s a Compassionate Use Program?
Let’s back up for a quick moment: Under current US Food and Drug Administration (FDA) regulations, if a company wants to conduct a clinical trial on a drug, it first needs to obtain regulatory approval to do so. It does this by submitting an investigational new drug (IND) application to FDA, which is essentially an exemption from federal law (which otherwise bans unapproved drugs from entering into interstate commerce) that allows a drug to be manufactured and investigated.
Under the terms of the IND, sponsors are tightly regulated, and are only able to use the drug on patients enrolled in the clinical trial. This is done to ensure that the drug is used safely, that the correct patients are enrolled in the trial, and that all side effects can be monitored.
OK—back to the original question: What is the compassionate use program?
Over the last two decades, FDA has moved to allow sponsors to expand access to their products, more commonly known as “early access programs” or “compassionate use exemptions.”
The intent of these programs is to allow FDA to permit companies to broaden access to investigational products while they’re still undergoing clinical trials.
For example, assume a drug is being tested for one type of cancer. You, however, have a different type of cancer that will almost certainly kill you, and all existing options have failed to treat the cancer. Your doctor believes that the drug being tested for the other type of cancer might be of some benefit.
Under normal IND regulations, FDA would be unlikely to approve research in this case because it would not advance any understanding of the drug’s safety or efficacy. The intent of the expanded access/compassionate use programs, however, is to allow patients who have the most to gain and the least to lose to access an investigational product.
Which Patients are Eligible for the Expanded Access Program?
Not all patients will be eligible for expanded access programs. Only patients with serious or immediately life-threatening diseases with no comparable or satisfactory therapeutic alternatives are eligible. Even then, the company must agree that to provide the drug to the patient and obtain FDA approval under one of several types of special INDs (discussed below).
The most important concept in expanded access programs is that the patient be aware of the risks he or she is undertaking, and that the company minimizes unnecessary risks to the extent possible. For that reason, FDA requires that all proposed uses first be approved by an Institutional Review Board (IRB), and that the patient (or the patient’s parent or guardian) sign an informed consent form.
FDA’s May 2013 guidance, Expanded Access to Investigational Drugs for Treatment Use—Questions and Answers, has more information. FDA has also published numerous guidance documents about expanded access and compassionate use programs.
How Does the Expanded Access Program Work in Regulatory Terms?
Expanded access works, in general, in one of two ways: Either a company with an experimental product creates a new clinical trial for a patient through the use of an IND, or it amends an existing clinical trial to add new types of participants through the use of a “protocol amendment.”
Once a company determines which approach it wants to take, it then needs to decide on how many patients it is willing to accommodate. There are four general types of expanded access INDs and protocols:
- Single Patient (Emergency Access): Used to grant access to a single patient who does not have time to obtain written permission from FDA
- Single Patient (Regular Access): Used to allow a single patient access to a trial
- Intermediate Size: Used for intermediate-sized patient populations
- Treatment: Used for large patient populations (i.e. widespread use).
Who Decides if a Patient Can Participate in a Compassionate Use Trial?
The company—not FDA. While FDA often works closely with companies to facilitate wider access to a drug, it is ultimately the sole choice of a company whether to grant expanded access to a drug or not.
Why Wouldn’t a Company Agree to Allow Access?
Companies and regulators alike have expressed some hesitancy about the program.
For companies, expanded access means letting products out of tightly controlled and heavily monitored environments, potentially subjecting the product to incorrect use and previously unknown adverse events, which would still need to be reported to FDA. Such incidents could potentially raise questions for regulators, thereby harming the chance of a product getting to market.
Further, some companies are concerned that expanded access programs could remove the incentive for patients to enroll in clinical trials meant to provide evidence for their drug’s full approval, thereby delaying its approval and harming other patients in the process.
“People at biotech companies therefore often must make emotionally difficult decisions when trying to balance an individual’s early access to a drug still in clinical trials against the company’s obligation to develop drugs for larger groups of patients and ensure these products gain regulatory approval as quickly as possible,” the trade group BIO wrote in March 2014. “In some cases, such early access programs could create a conflict between these two principles.”
Still other companies have cited the cost and staff resources necessary to administer compassionate use programs—a problem most evident in small biotechnology startups which do not yet have any income. Sometimes there are also concerns that there won’t be enough drug product available to supply both existing trials and new clinical trials, putting both groups of patients at risk.
To paraphrase BIO’s remarks again: While you’ll hear much about the one patient who isn’t obtaining access to a drug therapy, you’re unlikely to hear much about the thousands of patients who might have to wait several more months to gain access to an FDA-approved drug. And you’re probably not going to hear much about a company’s manufacturing woes or financial problems at the same frequency on social media either.
Regulators, too, are still wary of the program. “There’s this sense from patients that these are miracle drugs,” said Richard Klein, director of FDA’s Office of Special Health Issues, to the Wall Street Journal in October 2012. That feeling isn’t necessarily true. Even if the drug works-which is far from certain in early-stage clinical trials-the benefit might not amount to much. And worse, the drug may actually hasten a patient’s death, putting regulators in an uncomfortable position.
You Mentioned the Cost to Companies-Can They Charge Patients for Investigational Treatments?
Yes they can. Under a May 2013 guidance document, Charging for Investigational Drugs under an Investigational New Drug Application, FDA confirmed that companies may charge for expanded access treatments as long as they meet a four-part test:
- The drug must exhibit evidence of a clinical benefit.
- Data from the trial is essential to obtaining future approval for it.
- The trial could not be conducted without charging.
- The amount being charged is reasonable.
The sponsor may either charge the patient directly or the patient’s insurance company, but may only charge for the direct costs of providing the drug-its manufacturing, shipping, monitoring, third-party administration, etc.
Some companies may be wary of charging patients for the drug, however. According to a former FDA official involved in overseeing the compassionate use program, some companies believe that charging a “reasonable” amount may impact their ability to negotiate a higher sale price for the drug at a later date.
What Form Should my Doctor Fill out to Allow me to Participate in an Expanded Access Program?
On 4 February 2015, FDA announced it would dramatically streamline the process used by most physicians to enroll their patients into expanded access trials.
Doctors can now use the FDA Form 3926 to enroll a patient. The form, as outlined in FDA’s draft guidance document, Individual Patient Expanded Access Applications: Form FDA 3926, calls for doctors to submit the following eight pieces of information:
- Box 1: Patient’s initials (not the full name, to preserve confidentiality) and date of submission.
- Box 2: Clinical information, including indication, brief clinical history of the patient (age, gender, weight, allergies, diagnosis, prior therapy, response to prior therapy), and the rationale for requesting the proposed treatment, including an explanation of why the patient lacks other therapeutic options.
- Box 3: Treatment information, including the investigational drug’s name and treatment plan. This includes the planned dose, route and schedule of administration, planned duration of treatment, monitoring procedures, and planned modifications to the treatment plan in the event of toxicity.
- Box 4: Letter of authorization (LOA) obtained from the investigational drug’s manufacturer and attached to draft Form FDA 3926, when finalized. An LOA grants FDA the right to reference the application for information to satisfy submission requirements, such as a description of the manufacturing facility, chemistry, manufacturing and controls information, and pharmacology and toxicology information.
- Box 5: Physician’s qualification statement that specifies the medical school attended, year of graduation, medical specialty, state medical license number, current employment, and job title.
- Box 6: Physician’s name, address, and contact information, including the physical address, email address, telephone number(s), facsimile number, and IND number, if known. (Or IND number)
- Box 7: Request for authorization to use Form FDA 3926 for individual patient expanded access to comply with FDA’s requirements for submitting an individual patient expanded access IND.
- Box 8: Certification statement and signature of the physician certifying that treatment will not begin until 30 days after FDA receives the application unless the submitting physician receives earlier notification from FDA that the treatment may proceed; that the physician will obtain informed consent in compliance with FDA’s regulations in 21 CFR part 50; that IRB review of the expanded access use will be obtained in compliance with FDA’s regulations in 21 CFR part 56; and that in the case of an emergency request, treatment may begin without prior IRB approval provided the IRB is notified of the emergency treatment within 5 working days of treatment.
An old form, known as Form FDA 1571, called for physicians to submit 26 separate types of information and seven attachments, an FDA official said in a blog post explaining the change. In contrast, the new form requires just eight elements and a single attachment.
FDA expects the form to be able to be filled out in 45 minutes instead of 100 hours.
Why do I keep Hearing about Compassionate Use Programs?
Almost by definition, patients eligible for compassionate use programs are in danger of dying. For the patients and family members of those patients, access to experimental medicines can represent one last chance to fight against a disease or condition that will otherwise kill them or their loved ones.
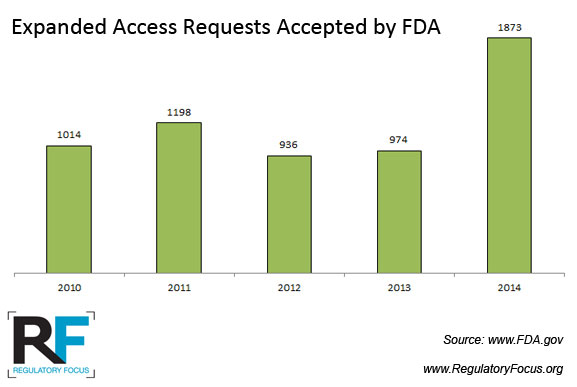
Interest in the program is also growing. According to the Wall Street Journal, participants in the program grew from around 1,000 patients nationwide in 2010 to more than 1,200 in 2012.
And in recent years, the fight to obtain those medicines has taken to social media time and again. In March 2014, for example, Chimerix was the subject of an intense lobbying campaign by the family and supporters of a boy named Josh Hardy. Hardy was suffering from a bacterial infection that had defied other treatments, and was seeking access to the company’s experimental drug, brincidofovir. While Chimerix initially refused to grant Hardy access to the drug, citing the prohibitive cost to the company, it eventually relented.
Similar cases pop up on nearly a daily basis. By way of example:
- Nick Auden, who had a 500,000-person petition seeking access to a Merck drug
- Darlene Gant, who sought access to Genentech’s pertuzumab
- Andrea Sloan, who sought BioMarin Pharmaceuticals’ BMN673
- Jack Fowler, who sought Shire’s SHP-609
Are Some States Working to Address This Issue as Well?
Yes. Several dozen states have introduced legislation intended to address various aspects of the compassionate use process. Most are primarily intended to shield doctors from being sued by patients, but others are more comprehensive.
Are Patients Ever Denied Participation in Proposed Expanded Access Programs?
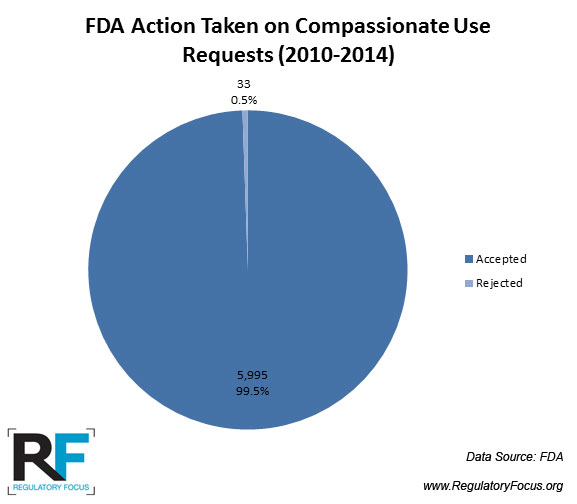
All the time, but rarely by FDA.
FDA data indicate that between 2010 and 2013, it rejected just 24 expanded access INDs, and no expanded access protocols were rejected. The majority of rejections were related single-patient emergency INDs.