Merck & Co’s Keytruda cancer immunotherapy is set to become the world’s top-selling drug by annual revenue in 2023, smashing sales records set by AbbVie’s inflammatory disease drug Humira, according to a new forecast.
According to the forecast from GlobalData, PD-1 class Keytruda (pembrolizumab) will earn US-based Merck & Co and Otsuka Holdings more than $22.2 billion by 2025.
Followed by Keytruda, Bristol-Myers Squibb and Pfizer’s Eliquis (apixaban) the novel oral anticoagulant will be the second best selling drug in the top 10, with annual sales of $18.7 billion by 2025 according to the forecast by GlobalData.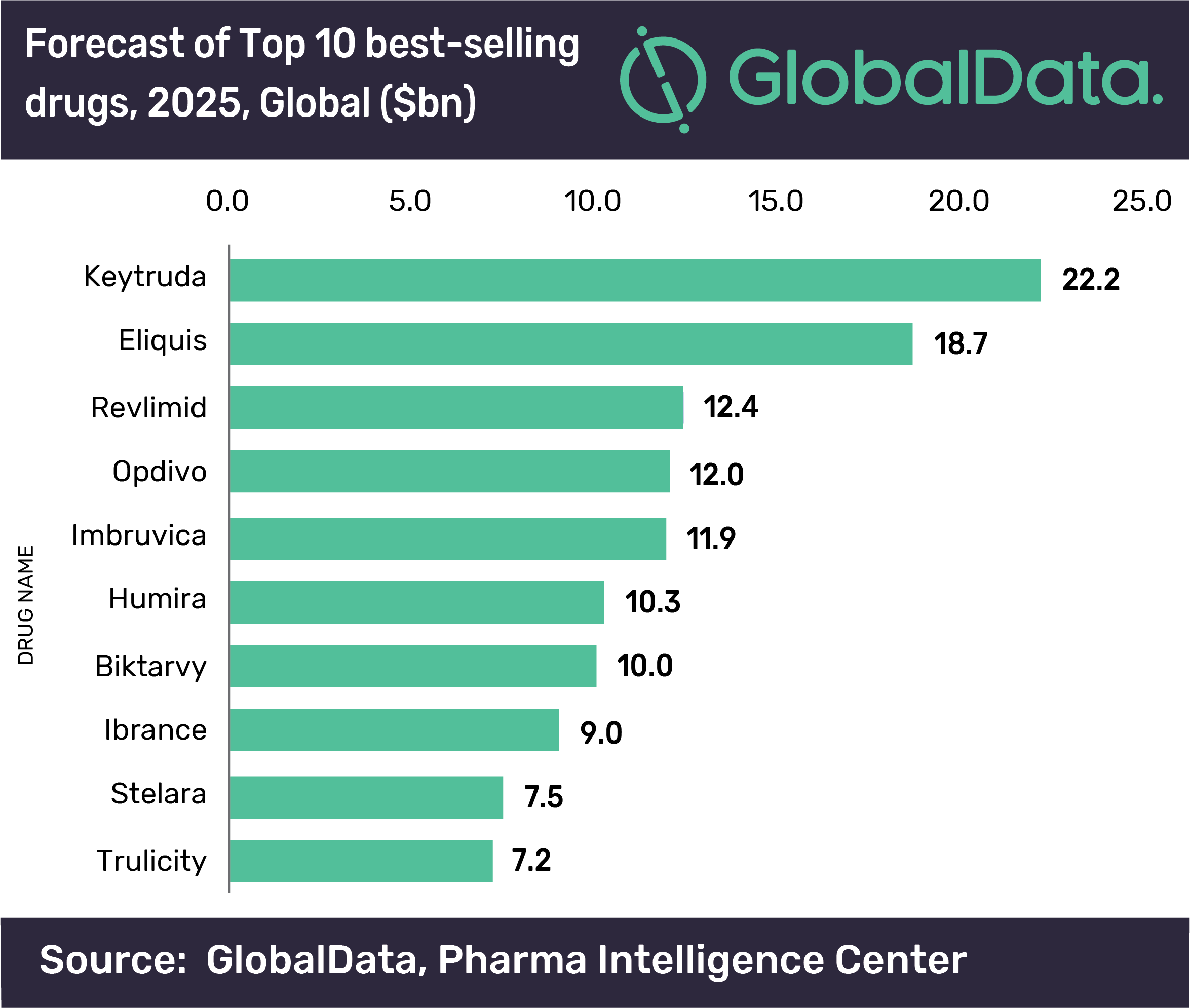
The figures mean that Keytruda will break sales records set last year by AbbVie’s Humira (adalimumab) inflammatory diseases drug, which peaked at just under $20 billion annually but is now being affected by patent expiries outside the US.
Like Humira, which has multiple inflammatory disease indications, Keytruda’s strength is in its versatility, as it has been approved for more than 20 oncology uses in the US, with many more likely to follow, as well as in new combinations with other drugs.
By 2025, Humira will have dropped down the list of 10 best selling drugs, following patent expiry in the US in 2023 when a gang of cheaper biosimilar rivals are set to hit the market.
According to GlobalData, Keytruda’s rival from Bristol-Myers Squibb, Opdivo (nivolumab) will be the fourth largest drug with annual sales of $12 billion, slightly behind Celgene’s Revlimid (lenalidomide) in third place on the list with $12.4 billion.
J&J and AbbVie’s Imbruvica (ibrutinib) comes in at fifth place in the forecast with sales of $11.9 billion.
Humira comes next with sales of $10.3 billion, followed by Gilead’s HIV drug Biktarvy ($10 bn), Pfizer’s breast cancer drug Ibrance ($9 billion), Johnson & Johnson’s Stelara for inflammatory diseases ($7.5 billion), with Eli Lilly’s Trulicity in tenth place with annual sales of $7.2 billion.