For more than a decade now, biotech and pharmaceutical companies have brought a new class of treatments – biosimilars – to markets around the world (from the EU to India to South Korea to the US), offering cost savings for some of the most expensive medicines, though even in 2016, decades after companies began their quest to develop biosimilars, they are still only just beginning to see widespread adoption.
What is a Biosimilar?
A biosimilar medicine (also sometimes known as a “follow-on biologic,” “subsequent-entry biologic” or “follow-on protein product”) is a medicine that is similar to another, already-authorized biologic medicine (including vaccines, blood and blood components, allergenics, somatic cells, gene therapies, tissues and recombinant therapeutic proteins).
As the US Food and Drug Administration (FDA) notes: Both biologics and biosimilars are isolated from a variety of natural sources – human, animal or microorganism – and can be composed of sugars, proteins, nucleic acids or complex combinations of these substances, or they may even be living entities, such as cells and tissues.
In the US and EU, the two biggest markets for biosimilars so far, a biosimilar must have no clinically meaningful differences in terms of quality, safety and effectiveness from the biologic that it is mirroring and is already authorized (that already authorized product is known as the reference product in the US).
Likewise, in both markets, companies have to carry out studies showing that the biosimilar is similar to the reference biologic and does not have any meaningful differences from the reference medicine in terms of quality, safety or efficacy.
However, as the European Medicines Agency (EMA) notes, data on a biosimilar’s reference biologic and the way it is used and made are already available, meaning the amount of data on safety and efficacy needed to win approval for a biosimilar is usually less than the amount needed to authorize the original biologic.
Why are Biosimilars Different From Generics?
Biosimilars and generics differ not only in size, stability and characterization, but also in how they are made, how they behave over time and their mode of action.
A lot of debate has been brewing about the comparisons between generics (which must be identical to their reference products) and biosimilars (which have a natural degree of variability, like with all different lots of the same biologic), with some media outlets calling biosimilars “copycats,” or “knockoffs,” though such euphemisms can oversimplify what is actually being developed.
Many in the pharmaceutical and biotech industries often to refer to generics as small molecule drugs and to biologics and biosimilars as large molecule drugs – meaning the molecules used as the active substances are either small (which for generics means they are synthesized by chemical reactions between different organic and/or inorganic compounds) or large (meaning they are composed of more than 1,000 amino acids and can be produced via modified cells of microorganisms, such as bacteria, yeast or in mammalian cell lines).
The Generics and Biosimilars Initiative (GaBI) offers a good example of the differences between a generic and biosimilar: An aspirin, which is a small molecule drug, measures just 180 daltons (a dalton is the standard unit used to indicate mass on an atomic or molecular scale) and has 21 atoms. That aspirin has a limited ability to initiate an immune response (how a body recognizes and defends itself against substances that appear foreign and harmful) and remains relatively stable over time. However, a biosimilar, which can be a monoclonal antibody or cell signaling protein, measures 150,000 daltons, contains 20,000 atoms, degrades over time and can generate a significant immune response.
As the Biotechnology Innovation Organization notes, differences between generic and biosimilar manufacturers are extensive. Biologics and biosimilars manufacturers must ensure consistency, quality and purity by ensuring that the manufacturing process remains substantially the same over time, whereas a small molecule manufacturer can change the manufacturing process extensively and analyze the finished product to establish that it is the same as before the manufacturing change.
Because of the complexity of biosimilars and the difficulty of manufacturing them, the cost of any given biosimilar is generally higher than the cost of a generic. And even as more biosimilars come to market and create more competition for costly biologics, the price difference between biosimilars and their reference products (biosimilars in the EU have typically been 15-30% cheaper) may never be as wide as the price difference between generics and their reference products.
Still, according to IMS, some EU countries are seeing a major impact from the introduction of biosimilars (though those cost savings may not be seen in the US until interchangeable biosimilars hit the market (see more in the Interchangeability section below)).
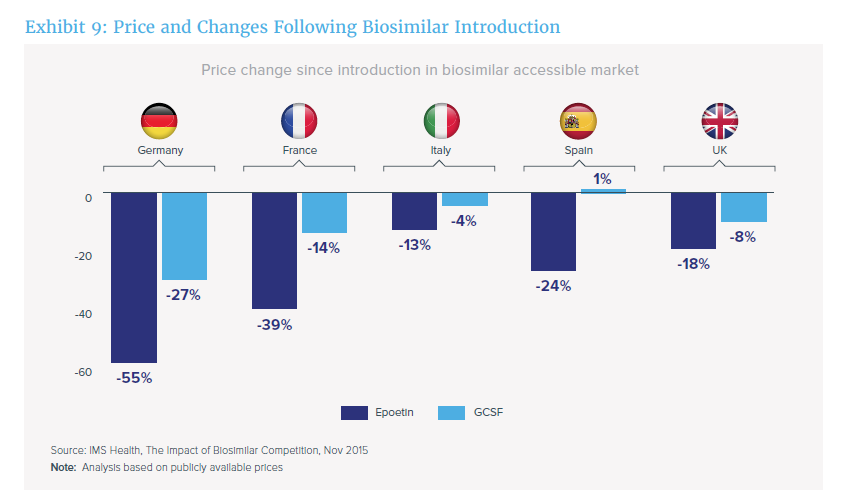
For the US, the potential cost savings is still to be determined, though current estimates will have to factor in a number of the issues outlined later on in this explainer.
IQVIA also offers a look at where the first biosimilars will become available first over the next ten years:
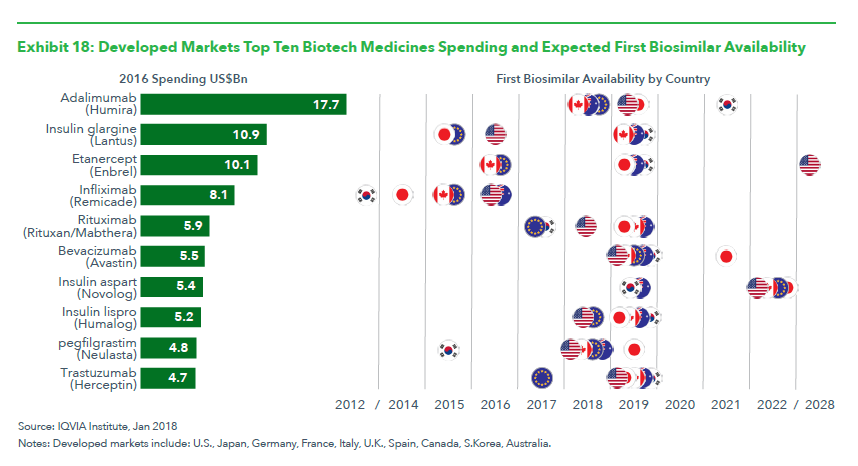
A number of other cost-related factors for US biosimilar savings were also outlined by CVS in a report (chart below).
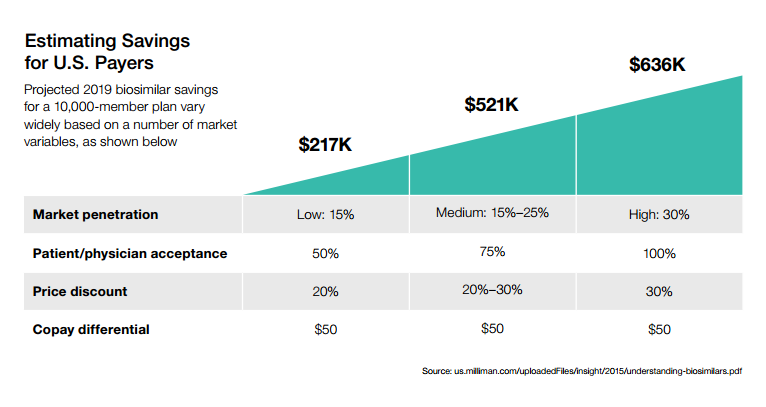
And as the pipeline of biosimilars continues to swell, more competition may mean lower-cost treatments (though many of these biosimilars have either been approved or submitted for approval since this chart was published – more on approvals below).
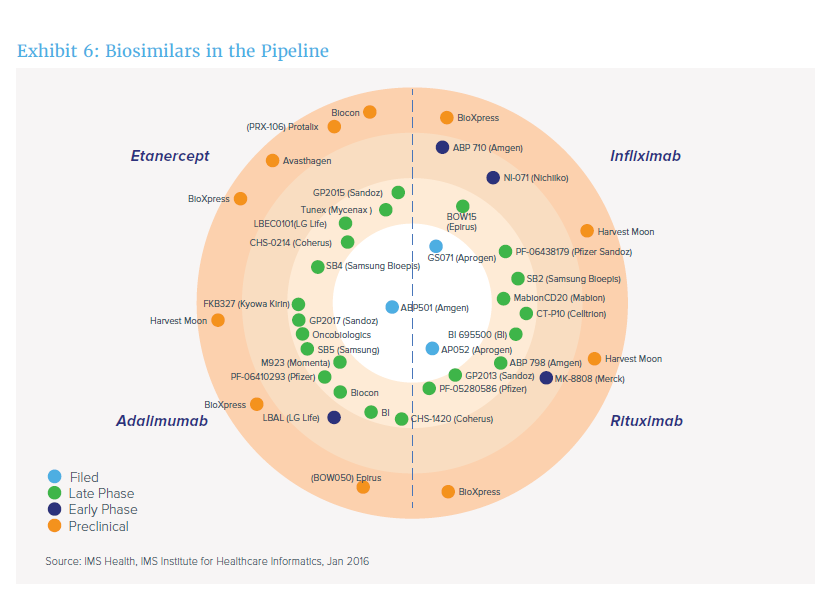
What Biosimilars are Approved and Where?
For the EU – which is the trendsetter – the experience with biosimilars (which can only be approved after the reference biologic has been on the market for 10 years – two years less than in the US, though in the US, a biosimilar can be filed four years after FDA approval of the reference biologic) has been relatively smooth, with more than 20 biosimilars approved between December 2006 and January 2018.
Remicade biosimilars now have 53% of EU market share when compared with their reference products, while Enbrel biosimilars see 29% market share and Rituxan (rituximab) biosimilars, which have yet to be approved in the US but are coming, have gained 13% of market share, according to Bernstein biotech analyst Ronny Gal.
In the US, however, FDA has authorized the use of nine biosimilars (and only three have actually come to market): Sandoz’s Zarxio (filgrastim-sndz), which is similar to Amgen’s Neupogen, competition for Enbrel with Erelzi (etanercept-szzs), which has yet to hit the US market and has patent protection through 2029; for Avastin with Mvasi (bevacizumab-awwb), which may not come to market in the US until early 2019; for Humira with Amjevita (adalimumab-atto) and Cyltezo (adalimumab-adbm) and for Herceptin with Ogivri (trastuzumab-dkst), which was approved in December 2017, though the launch date remains unknown. Pfizer’s Ixifi (infliximab-qbtx) was also approved in December 2017, though the company said it will not launch the product.
Remicade also has two biosimilar competitors approved by FDA – Samsung Bioepis’ Renflexis (infliximab-adba) and Celltrion and Pfizer’s Inflectra (infliximab-dyyb), both of which have launched, but uptake has been modest, barely scraping 5% of the market. The only other biosimilar to launch is Zarxio (filgrastim-sndz).
FDA is likely to approve more in the coming years, and the term biosimilar is tricky in the US as some approved follow-on biologic products, like insulins, are sometimes considered biosimilars, though FDA has said that in some cases, like with Eli Lilly’s Basalgar, they are not biosimilars as they are approved via a 505(b)2 application that relied partly on the application for Sanofi’s Lantus.
As of January 2018, 60 biosimilars were enrolled in the FDA’s biosimilar development program, and FDA has received meeting requests to discuss the development of biosimilars for 27 different reference biologics.
In India, which according to GaBI is actually ahead of the EU in terms of number of approvals, though the approval standards are not the same, more than 60 biosimilars have been approved since 2000. Guidance from the Central Drugs Standard Control Organization was first issued in 2012 and then updated in March 2016.
Japan’s Ministry for Health, Labour and Welfare (MHLW) has approved nine biosimilars (which includes insulins) between 2009 and 2016, according to GaBI, and released industry guidance on biosimilars back in 2009 (for more perspective on Japan’s experience with biosimilars, check out this article in The Lancet from officials from Japan’s Pharmaceuticals and Medical Devices Agency).
South Korea’s Ministry of Food and Drug Safety (MFDS), meanwhile, has approved six biosimilars between 2012 and 2015, and also published guidance back in 2009, relying on EU, Japanese and WHO biosimilar guidance.
Approval Pathways
In the US, a Biologics License Application (BLA) is a request for permission to introduce, or deliver for introduction, a biologic into interstate commerce. The BLA is regulated under 21 CFR 600 – 680 (Form 356h specifies the requirements for a BLA).
Under the Biologics Price Competition and Innovation Act of 2009 (BPCIA), enacted in March 2010, as part of the Patient Protection and Affordable Care Act (better known as Obamacare), a proposed biologic that is biosimilar to an FDA-approved biologic (the reference product) can use the abbreviated approval pathway under section 351(k) of the Public Health Service Act(PHS Act).
FDA’s biosimilar lead Leah Christl explained to Focus in June 2016: “The data packages differ between originator and biosimilar marketing applications, but the standard for approval of originator products under 351(a) and biosimilar products under 351(k) of the PHS Act is the same in that both must demonstrate that they are safe, pure, and potent for the approved conditions of use.”
In addition, she said:
- The sponsor of the proposed biosimilar product does not have to independently establish the safety and effectiveness of its product; it establishes this through a demonstration of biosimilarity to the reference product.
- The sponsor of a proposed biologic demonstrated to be biosimilar to a reference product can rely on FDA’s previous finding of safety, purity, and potency (i.e., safety and effectiveness) for the reference product. “The ability to rely on certain existing scientific knowledge about the reference product to support the safety, purity, and potency (i.e., safety and effectiveness) of the biosimilar product allows for a potentially shorter and less costly drug development program. This is what is meant by an abbreviated approval pathway.”
- The abbreviated pathway is not a lesser standard for approval.
- The data packages differ between originator (351(a) BLA) and biosimilar (351(k) BLA) marketing applications, but the standard for approval of originator and biosimilar products “is the same in that both must demonstrate that they are safe, pure, and potent for the approved conditions of use.
- “The data package required for approval of a biosimilar product is quite extensive; it is the approval pathway that is abbreviated.”
As far as industry concerns with FDA’s interpretation of the BPCIA, Sanofi, Mylan, Novo Nordisk, and industry groups PhRMA and the Biosimilars Council in May 2016 called on FDA to amend its interpretation of the “deemed to be a license” provision of the Act as some are saying the current draft guidance could halt biosimilar development for a prolonged period.
In addition to the 351(k) pathway, the Federal Food, Drug, and Cosmetic Act (the FD&C Act), as amended by the Biosimilar User Fee Act of 2012 (BsUFA), FDA can assess and collect fees for biosimilars from October 2012 through September 2017. The user fee program was reauthorized in August 2017 and will now extend through 2022 with additional fees.
FDA dedicates these fees (the latest of which can be found here) to expediting the review process for biosimilars.
On the EU end, a pathway for approving biosimilars has been in place since 2003.
“The main part of the evaluation is a comparison of the biosimilar with its reference medicine to show that there are no significant differences between them,” EMA says. “The relevant regulatory authority applies stringent criteria in their evaluation of the studies comparing the quality, safety and effectiveness of the two medicines. The studies on quality include comprehensive comparisons of the structure and biological activity of their active substances, while the studies on safety and effectiveness should show that there are no significant differences in their benefits and risks, including the risk of immune reactions.”
And like with the US, because the reference medicine has been authorized in the EU for a decade (12 years in the US) and its clinical benefit is well established, some studies carried out with the reference medicine may not need to be reproduced, EMA says.
Names
In the EU, biosimilars use a unique proprietary name but the same nonproprietary name (also known as an international nonproprietary name or INN) as their reference product, like with generics. For example, Biogen and Samsung’s Benepali (approved in the EU in January 2016) is biosimilar to Amgen’s Enbrel, and both use the non-proprietary name of etanercept.
In the US, however, biosimilar names have stirred up controversy, mostly because of a requirement (from naming guidance released in August 2015) to add a four-letter suffix at the end of a biosimilar’s nonproprietary name.
Industry groups commenting on the guidance, including the Biosimilars Forum, PhRMA and BIO, supported FDA’s proposal, but called for the agency to use “meaningful” and “distinguishable” suffixes linked to the license holder’s name, just like what was used for the first US-approved biosimilar, known as Zarxio (filgrastim-sndz).
BIO and PhRMA are also calling for biosimilar labels to describe the basis of approval for each indication with relevant data for the reference product and biosimilar.
However, the Biosimilars Council opposed FDA’s proposal, claiming that “adding a suffix … is unnecessary and does not achieve the Agency’s stated goals as it will lead to confusion among prescribers and patients.” Its comment goes on to argue that these products “have other names … for distinct recognition; including a brand name, company name, a lot number and a national drug code (NDC) number that readily distinguish [them] from other products.”
Insurers such as the Kaiser Foundation, CVS Health and America’s Health Insurance Plans (AHIP) fell in line with the Biosimilars Council’s stance, opposing the use of a suffix in favor of other measures to distinguish biosimilars for pharmacovigilance purposes.
The suffix is meant to help better track the safety and use of biosimilars, though alongside that guidance, FDA also proposed a rule to designate the proper names of six already approved biologics (which also are expected to get new names).
The products include filgrastim, pegfilgrastim, infliximab and epoetin alfa, each of which is a reference product for “an approved or publicly disclosed section 351(k) application,” a related biological product, tbo-filgrastim, and a biosimilar, filgrastim-sndz.
The rule proposes assigning a suffix to each of the six products, or replacing the product’s previous suffix (or prefix, in the case of tbo-filgrastim).
For example, Amgen’s Neupogen (filgrastim) would be changed to “filgrastim-jcwp,” while Sandoz’ Zarxio (filgrastim-sndz) would be renamed “filgrastim-bflm.” For the biosimilar Granix (tbo-filgrastim), FDA has proposed removing the prefix “tbo” and adding a new suffix, “filgrastim-vkzt.” The agency says it believes changing the names of these products will help “avoid confusion regarding these products relationships to one another.”
Additionally, by using only suffixes, related or similar products can be “grouped together in electronic databases,” while remaining distinguishable.
The agency also says it is considering alternative names for these specific products that would still use a four letter suffix, but where the suffix is based on the product license holder’s name, such as “amgn” for Amgen, or “jnsn” for Janssen.
The issue has been so controversial that FDA in June 2016 released a document calling on companies to select 10 suffixesdevoid of meaning for biosimilar and biologic names, though the agency then withdrew that document three weeks later and FDA’s Christl has since clarified that companies should submit one to three suffixes.
WHO, meanwhile, has drafted a proposal to use what it calls a “biological qualifier” to further distinguish biosimilars from their reference products.
Both BIO and PhRMA have urged FDA to engage with WHO and other stakeholders to discuss harmonization, and the Federal Trade Commission (FTC) and USP have offered support for WHO’s approach, with the FTC saying: “unlike FDA’s proposed suffix, which would be part of the nonproprietary name, the [biological qualifier] would be separate from the nonproprietary name in pharmacy databases.”
Labels
In March 2016, FDA unveiled draft guidance on biosimilar labels, which will rely heavily on their reference products’ labels, though the biologics industry will likely be happy that the labels must make certain clarifications about the biosimilar and reference product.
FDA says that like with generics, biosimilar labeling should include a description of the clinical data that supported safety and efficacy of the reference product.
But in circumstances where a biosimilar is not approved for the same indications as its reference product, unlike with generic drugs, FDA notes that “it may be necessary to include information in the biosimilar product labeling relating to an indication(s) for which the biosimilar product applicant is not seeking licensure, in order to help ensure safe use (e.g., when safety information in the reference product labeling is related to use of the product and is not specific to a particular approved indication(s) or when information specific to only the biosimilar product’s indication(s) cannot be easily extracted).”
And the label should be written in a manner that does not imply that the biosimilar product is approved for a reference product indication(s) or use(s) that has not been approved for the biosimilar product, the draft guidance says.
“The overall risk-benefit profile of the reference product is relevant to the biosimilar product, even if a particular serious adverse reaction or other risk included in the reference product labeling may not have been reported with the biosimilar product at the time of licensure,” FDA adds.
FDA also offers illustrative examples with a fictional reference product JUNEXANT (replicamab-hjxf) and a fictional biosimilar product NEXSYMEO (replicamab-cznm).
In labeling sections where the risk applies to both the biosimilar product and the reference product (e.g., BOXED WARNING, CONTRAINDICATIONS, WARNINGS AND PRECAUTIONS, ADVERSE REACTIONS (Postmarketing Experience)), it would be appropriate to use the core name of the reference product followed by the word “products” (i.e., replicamab products) to convey, for example, that a risk or other information necessary for the safe use of the product applies to both the biosimilar and the reference product.
In sections where the information described is specific to the biosimilar product — i.e. INDICATIONS AND USAGE, DOSAGE AND ADMINISTRATION, DOSAGE FORMS AND STRENGTHS, DESCRIPTION, and HOW SUPPLIED/STORAGE AND HANDLING – FDA says:
- For directive statements and recommendations for preventing, monitoring, managing, or mitigating risks (e.g., “Discontinue NEXSYMEO in patients with [adverse reaction]”) — Such statements are typically included in, but are not limited to, the BOXED WARNING, CONTRAINDICATIONS, WARNINGS AND PRECAUTIONS, and DRUG INTERACTIONS sections.
- When clinical studies or data derived from studies with the reference product are described in the biosimilar labeling, the reference product’s proper name should be used. This information would typically be included in sections such as, but not limited to, ADVERSE REACTIONS (Clinical Trials Experience) and CLINICAL STUDIES.

Interchangeability
The biggest difference between the EU and US biosimilar markets is “interchangeability,” a yet-to-be-fully-explained term used to describe biosimilars in the US for which additional studies were conducted to evaluate multiple switches between the reference product and biosimilar. Though no interchangeable biosimilars have been approved in the US, FDA has said the first may come to market in the next two years. The term designates a particular class of biosimilars that can be automatically substituted at the pharmacy level.
In January 2017, FDA issued draft guidance on interchangeability, with FDA asking for a multiple switch study with pharmacokinetic endpoints, that the reference product used as the comparator be sourced from the US, and that extensive human factor studies be conducted with the device.Many have commented on the draft interchangeability guidance, though Boehringer Ingelheim is the only company to have publicly disclosed that it is conducting a clinical study designed to meet the interchangeability requirements outlined in the draft guidance.
The term does not exist in the EU as EMA explains that its evaluations do not include recommendations on whether a biosimilar should be used interchangeably with its reference medicine, and the decisions on switching from one biological medicine to another are referred by EMA to doctors and pharmacists.
Drug, biologic and biosimilar companies, meanwhile, are debating (in comments on FDA’s draft guidance on biosimilar labeling) over whether to include a biosimilar’s interchangeability status on each new product’s label.
State Legislation
Although the BPCIA established the pathway whereby biosimilars can reach the market in the US, states (heavily influenced by lobbyists from the biopharma industry) have taken the matter of substituting biologics for biosimilars into their own hands.
Twenty-four states (the most recent of which was Pennsylvania in July 2016) and Puerto Rico now have enacted laws creating a patchwork of regulations governing how biosimilars are dispensed, even before biosimilars have hit the market en masse in the US and before FDA has explained what interchangeability means.
The laws generally require pharmacists to only be allowed to substitute a biosimilar for its reference product if FDA has determined the biosimilar to be interchangeable, and if the prescriber does not prohibit such a substitution, if the pharmacy informs the patient of the substitution and if the pharmacy retains a record of the biosimilar dispensed, and within five business days of dispensing a biosimilar, the pharmacist records what specific product was provided to what patient, including the name of the product and the manufacturer.