
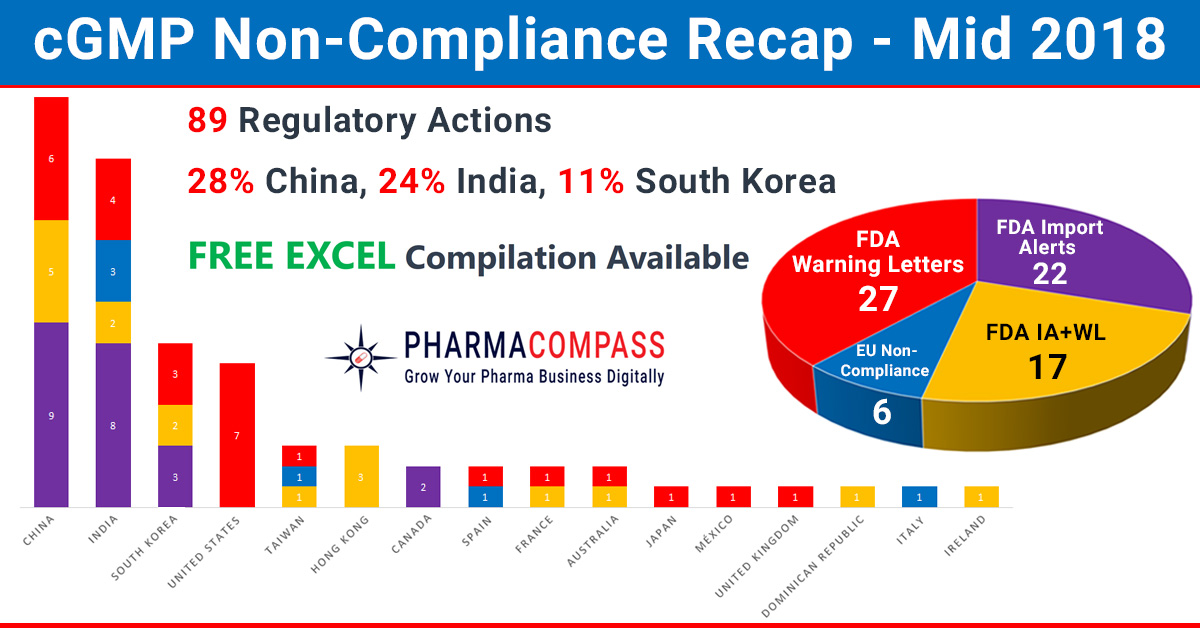
In our mid-2018 compliance review, we look at inspection challenges faced by companies across the world. In the first half of this year, manufacturing compliance challenges dominated headlines. But we also saw shortcomings at major pharmaceutical companies like Pfizer, Bayer and Akorngenerate news.
While China, India and the US continued to be the top three countries where regulators uncovered compliance issues, this year has also seen the FDA take action against many South Korean companies. The European authorities found concerns in India, Taiwan, Italy and Spain. However, there were no non-compliance reports issued to firms in China until the end of June 2018.
While data-integrity violations and a failure to thoroughly investigate deviations continued to remain a major concern for inspectors, this year the real concern emanated from the supply of product to market (which had the potential to impact product quality or patient safety).
China: API with a cancerous impurity, vaccine scandal and data-integrity woes
The most recent regulatory non-compliance issue pertains to the European Medicines Agency (EMA) raising concern over the active pharmaceutical ingredient (API) valsartan supplied by China’s Zhejiang Huahai Pharmaceuticals.
The concern was the impurity — nitrosodimethylamine (or NDMA) — detected by the company in their valsartan API. NDMA is classified as a probable human carcinogen and its presence was unexpected as it was not detected by routine tests carried out by Zhejiang Huahai.
Zhejiang Huahai sold over US$ 50 million of the API in 2017 and supplies to most major manufacturers producing valsartan medicines available in the EU and United States.
While a review is underway, national authorities across the EU, US and Asia are recalling medicines containing valsartan supplied by Zhejiang Huahai.
Vaccine scandal: A major vaccination scandal has sparked off a huge outcry in China as vaccine maker Changsheng Biotechnology was found to have falsified production data for its rabies vaccine.
Changchun Changsheng Bio-tech Co, in Changchun, reported serious irregularities, including fabricating production records in the manufacture of rabies vaccines for human use, during an inspection by the State Drug Administration, China FDA said in a statement.
Although there has been no evidence of harm from the vaccine, the firm has been ordered to halt production and recall rabies vaccines. And Chinese Premier Li Keqiang has urged severe punishment for the people involved, saying the incident had “crossed a moral line”.
Data-integrity violations: This year, the FDA also posted the warning letter issued to Henan Lihua Pharmaceutical in China, a company that produces steroid APIs like hydrocortisone and prednisone.
The warning letter highlighted data integrity concerns that landed Henan on FDA’s import alert list in March 2018.
During the inspection, the FDA investigator observed numerous blank batch manufacturing records in an open cabinet in the firm’s manufacturing workshop office.
Among these was multiple blank, product release forms marked with a red quality assurance release stamp stating ‘Permitted to Leave [the] Factory’.
The FDA also posted a warning letter issued to Jilin Shulan Synthetic Pharmaceutical, a manufacturer of caffeine API in China. The letter revealed flagrant data-integrity violations.
Another warning letter was issued by the FDA to API manufacturer Lijiang Yinghua Biochemical and Pharmaceutical, following an October 2017 inspection.
United States: Drug shortages due to Pfizer’s manufacturing problems
Drug major Pfizer’s production problems continued to make headlines this year. An article in Fortune put the blame on Pfizer’s much-touted US$ 17 billion acquisition of Hospira in 2015 for turning the United States’ chronic drug shortage into a full-blown crisis.
According to the article, as of May 11 this year, Pfizer — which is the world’s largest maker of sterile injectable drugs — had 370 products that are depleted or in limited supply, 102 of which the company has indicated will not be available until 2019.
“The simple answer to why America currently has so many shortages of generic sterile injectable drugs: America’s leading manufacturer of generic sterile injectable drugs hasn’t been making them,” the article said.
Mylan’s flagship product EpiPen is also likely to face shortages due to problems at Pfizer. Although Mylan owns the rights to the EpiPen, it subcontracts manufacturing of the auto-injector to Meridian Medical Technologies, a division of Pfizer.
While Mylan is putting pressure on Pfizer to do more to tackle shortages of this life-saving medicine, Pfizer has struggled to meet demand for the EpiPen and the FDA had put the medicine on its official shortages list.
In September last year, the FDA had issued a warning letter to Meridian Medical Technologies over serious component and product failures that had been associated with patient deaths.
Pfizer’s troubles are far from over as an FDA inspection of an ex-Hospira sterile manufacturing facility in India resulted in the issuance of a 32 page Form 483. The same facility was issued a warning letter by the FDA in 2013.
Germany: FDA highlights contamination, data-integrity concerns at Bayer facility
In a shocking warning letter issued by the FDA to Bayer Pharma’s finished pharmaceuticals manufacturing facility located in Leverkusen, Germany, investigators found compliance shortcomings ranging from concerns over data-integrity to serious product contamination problems.
While reviewing a drug product manufacturing operation, FDA investigators found residue on equipment which seemed most likely from a drug product that had been previously processed in the same room.
When Bayer tested the samples of the tablets being produced to “assess the potential of cross-contamination”, the testing confirmed contamination of the previously processed product inside the tablets which resulted in a recall of several lots of drug products.
India: Data-integrity violations, invalidation of OOS results continue
Alkem has ‘no quality control unit’: After eight days of inspecting Alkem Laboratories’ finished formulation facility in India in March 2018, the FDA investigators concluded — “there is no quality control unit”.
Alkem’s head of quality control (QC) and quality assurance (QA) confirmed out-of-specification (OOS) results for the assay for a batch of tablets. However, the company did not recall the product, which was distributed in the US market.
Less than three weeks before the inspection, the “firm’s QC department deleted two-thousand one hundred one (2,101) files” on its computer network.
Alembic invalidated OOS results: In the seven days that the FDA investigator — Jessica L Pressley — spent at Alembic Pharmaceuticals’ oral solid dosage manufacturing facility in Tajpura, Gujarat, she uncovered that the firm invalidated 131 of the 140 OOS results (an invalidation rate of 94 percent) for products marketed in the US.
The firm attributed the invalidation to analyst errors. In 2017, the invalidation rate was 91 percent.
The Form 483 shares a concern that the “OOS results that were invalidated by the firm’s QC unit were without rationale and supporting documentation.”
Alchymars falsified lab data: A September 2017 inspection by the USFDA at Alchymars ICM SM Private Limited in India uncovered that the firm “was falsifying laboratory data”. During the inspection, the FDA investigator found that an analyst reported far fewer colony-forming units (CFU) in a water sample than those observed on the plate by the investigator.
The FDA raised serious concerns as Alchymars uses the water to manufacture APIs intended for use in sterile injectable dosage form drug products.
Alchymars is part of a group of companies and the factory is controlled by Trifarma in Italy, a company which was cited by the FDA for data-integrity violations in 2014.
South Korea: Teva’s potential blockbuster gets delayed due to problems at Celltrion
As Korea emerges as a force to reckon with in the emerging world of biosimilars, the USFDA’s issuance of a warning letter to Celltrion (a major manufacturer of biosimilars that has also partnered with Pfizer for commercialization in the United States) came as a major setback.
In an inspection conducted by the FDA from May 22 to June 2, 2017, the investigators raised concerns over multiple poor aseptic practices during the set-up and filling operations.
The warning letter highlights an example where during the aseptic filling of vials, an operator used restricted access barrier system (RABS) to remove a jammed stopper by reaching over exposed sterile stoppers in the stopper bowl. The RABS disrupted the unidirectional airflow over the stopper bowl, creating a risk for microbial contamination.
After the operator removed the jammed stopper, the filling line was restarted, but the affected stoppers were not cleared.
At Celltrion, the FDA raised concern over 140 complaints received between October 2015 to May 2017, which were identified to have occurred because of vial stoppers.
The deficiencies at Celltrion impacted Teva as the Korean company is the main API supplier for Teva’s migraine drug fremanezumab.
Teva confirmed that the USFDA had extended the goal date of the Biologics License Application (BLA) for fremanezumab. The Prescription Drug User Fee Act (PDUFA) action date for fremanezumab is currently set for September 16, 2018.
The Celltrion warning letter was followed by an announcement by the US-based Evolusthat a USFDA pre-approval inspection of Daewoong Pharmaceutical’s plant in South Korea, where a botox biosimilar is being produced, resulted in 10 observations.
Back in 2013, Daewoong had inked a contract with Evolus to export DWP-450 (a botulinum neurotoxin candidate), which was expected to be released in the US market around 2017-18.
While Daewoong said it expects “no significant further actions”, Evolus’ SEC filing highlights that “any failure to adequately resolve the FDA’s observations at the Daewoong facility would likely cause FDA approval of DWP-450 to be delayed or denied”.
In May, the FDA declined to approve Evolus’ Botox rival citing deficiencies related to the chemistry and manufacturing of its potential treatment for frown lines.
Hey! I know this is kinda off topic but I’d figured I’d ask.
Would you be interested in exchanging links or maybe guest writing a
blog post or vice-versa? My blog discusses a lot of the same
subjects as yours and I believe we could greatly benefit
from each other. If you happen to be interested feel free to shoot me an email.
I look forward to hearing from you! Excellent blog by the way!