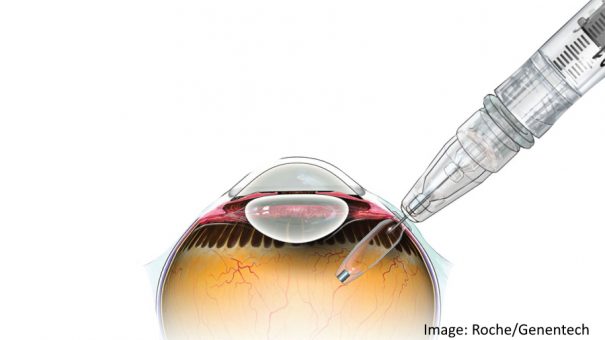
Roche’s Genentech unit has won FDA approval for Susvimo, an implant for eyesight-robbing disease wet age-related macular degeneration (AMD) that gives patients an alternative to regular injections into the eye.
Susvimo has the same active ingredient as Roche’s big-selling therapy Lucentis (ranibizumab), a VEGF inhibitor administered once a month that has been approved by the FDA since 2006 for wet AMD.
The port delivery system (PDS) implant is about the size of a grain of rice, is refillable and delivers the drug continuously for at least six months, so patients can manage wet AMD with just two treatments a year.
The FDA has approved Susvimo for the treatment of people with wet AMD who have previously responded to at least two anti-VEGF drugs such as Lucentis or Bayer and Regeneron’s rival Eylea (aflibercept).
The green light comes on the back of the phase 3 ARCHWAY trial which showed that Susvimo was equivalent to monthly Lucentis injections at maintaining vision in patients over 40 weeks, with 98% of patients lasting six months before needing a refill.
Additional Lucentis injections can be administered on top of Susvimo if needed, but just 1.6% of patients in the clinical trial needed the additional dosing.
In general, the implant was better tolerated than the regular Lucentis injections, although it was associated with a three-fold higher rate of endophthalmitis, an inflammation of the intraocular fluids.
In the 418-patient trial there were four cases, one of which resulted in permanent, severe vision loss, while the other three were treated and vision was restored to the level it was before implantation.
In a patient-reported outcome (PRO) survey, Susvimo recipients reported a strong preference for the implant over intraocular injections.
Roche said Susvimo would be priced at $9,250 for the first six months, with refills set at $8,000, which compares with a list price of around $1,230 for a monthly dose of Lucentis.
Cheaper biosimilar versions of Lucentis from Samsung Bioepis/Biogen, Hospira and Formycon/Bioeq are however due to enter the US market shortly.
Roche and Novartis share rights to Lucentis, with Roche selling the drug in the US and Novartis selling it in Europe and other markets.
Last year, Roche booked around $1.5 billion in US sales, while Novartis’ sales of the drug topped $1.9 billion, although the market leader remains Eylea which made more than $8 billion – thanks in part to more flexible dosing.
Roche and Novartis have gone in different directions as they prepare to defend their wet AMD franchises. While Roche has placed its faith in Susvimo, Novartis focused on the development of a longer-acting VEGF antibody – Beovu (brolucizumab) – which is dosed every two or three months.
Beovu has struggled to gather momentum since its launch because of concerns about its safety, with second quarter sales only reaching $47 million.
The jury is also out on the prospects with Susvimo, with some analysts concerned that the endophthalmitis issue may hinder early take-up, making it hard to displace Eylea from its current dominant position – particular as cut-price biosimilars to Bayer and Regeneron’s drug are due in the US in 2023.
Roche has another iron in the wet AMD fire however with faricimab, a bispecific antibody targeting both VEGF and Ang-2 that only needs to be injected every four months and has shown impressive efficacy in clinical trials. The FDA started a priority review of the drug in July.