Before the onset of the coronavirus pandemic, leading organizations in clinical research had been gradually discovering, experimenting with, and implementing remote technology. The pandemic has catapulted their transformation agendas forward.
This acceleration challenges the industry by shortening often lengthy vendor evaluation processes, uprooting existing cross-departmental workflows, and requiring swift and meticulous compliance and regulatory rework. Yet there are opportunities for organizations that face this change head on: avoiding costly study delays, continuing patient participation in critical chronic disease studies, accessing higher levels of study oversight and compliance adherence, and more.
I offer a look at how the technology landscape for clinical research accelerated in 2020, raise some risks this rapid acceleration poses, and offer suggestions for how leaders in clinical research and operations can navigate these changes for successful outcomes.
The shifting landscape
Over the past decade, clinical operations executives have been slowly adopting new technologies to reduce their bottom lines and increase the efficiency of clinical research. Covid-19 has accelerated their transformation timelines for implementing new technologies and approaches. Former technology infrastructure estimates for sites, sponsors, and clinical research organizations (CROs) for 2025 have now been pulled forward to 2021.
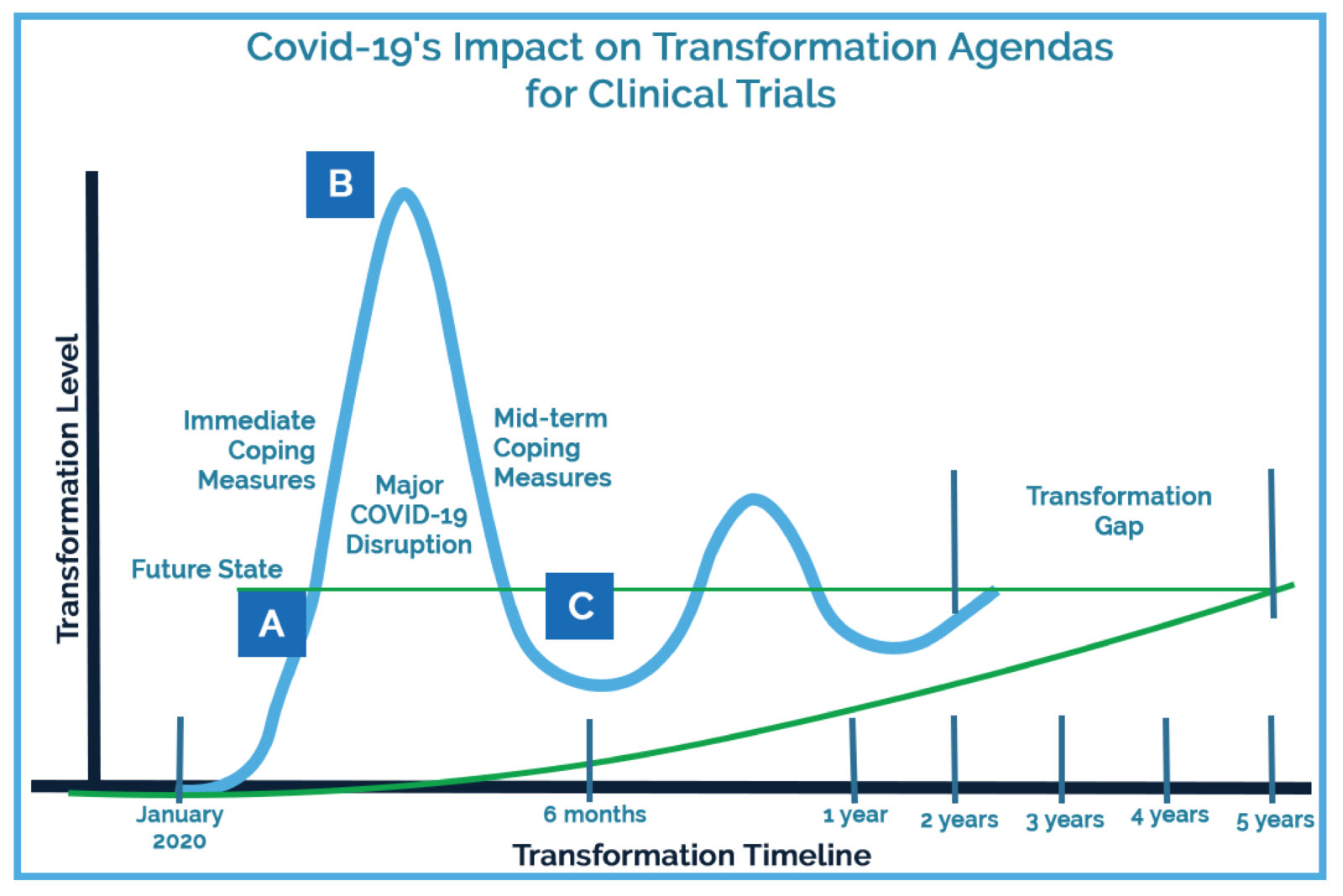
Part of the dramatic industry shift can be attributed to protocols put in place by research sites to ensure the safety of both patients and research staff as the pandemic began expanding globally.
These protocols include:
Although these measures helped reduce the spread of Covid-19, they required sites, sponsors, and CROs to reimagine how to safely conduct clinical trials in remote, technology-based environments. This was no small task, as adopting new technology at any research organization requires administrative, regulatory, and process changes.
Standard operating procedures and protocols that formerly addressed manual and paper processes had to be adjusted to include remote, technology-based methods. This shift also required clinical research coordinators, clinical research associates, and other research staff to learn and integrate technology into their workflows.
As these changes take hold, the barrier to future technology adoption and an increase in technology-based skills among research staff continues to lower, further accelerating the shift to permanent remote-capable technology processes in clinical research.
Risks of rapid acceleration
Adapting quickly in order to continue vital clinical research on treatments for Covid-19 and other diseases means evaluating and adopting technology platforms to accommodate new remote needs and keep research moving forward. The vendor evaluation process has become shorter than ever, as have adoption timelines, leaving room for error.
Increased vendor selection hurdles. The market is being flooded with new technology platforms to meet remote research demands. This influx can overwhelm research teams and add complexity to vendor selection by increasing the platforms and capabilities to sift through. This complexity increases the risk that sites, sponsors, and CROs will select platforms that don’t appropriately address their needs.
Shortened evaluation timelines. Research organizations typically form vendor evaluation teams that include members from various departments that will be affected by the technology. Under normal circumstances, vendor evaluations can take anywhere from six to 18 months, which includes time for research teams to test a platform’s workflows in a live environment. Accelerated transformation timelines are shortening this evaluation period, which may cause temporary resource strain and intensified decision-making.
Increased risk of system integration failures. Verifying that a new technology can integrate with current systems is an essential step for building robust, remote-ready technology infrastructures for clinical research. Vendor evaluation teams typically complete thorough investigations of the existing technology infrastructures of all involved research partners — sites, sponsors, and CROs — to avoid adopting technology systems that are redundant or incompatible with existing platforms. Accelerated transformation timelines hurry this infrastructure investigation, increasing the risk of overlooking current systems, miscalculating integration capabilities, producing integration failures, and creating inefficient or redundant workflows.
Rushed system testing and validation. Many organizations, especially large sponsors and CROs, now require a security evaluation to assess a technology platform’s regulatory and compliance abilities. This is helpful and often recommended to mitigate any compliance risk and verify system security and privacy. Yet to be effective, these evaluations require vendor cooperation, resource allocation, and timely responses. As vendor evaluation timelines shorten, rapid system testing and validation by research organizations increase the potential to overlook security or compliance risks identified in the questionnaire.