As Europe has been significantly impacted by the Covid-19 pandemic, a substantial amount of clinical research is currently focused on identifying effective therapies and vaccines. GlobalData analysed the Covid-19 clinical trials landscape in Europe with a view to identifying key trial-related trends. The greatest proportion of European Covid-19 clinical trials are in the Phase II stage of development (43.3%), narrowly outnumbering Phase III trials (38.4%). Phase IV clinical trials (12.6%) outnumber Phase I studies, which account for the smallest proportion of trials (5.6%).
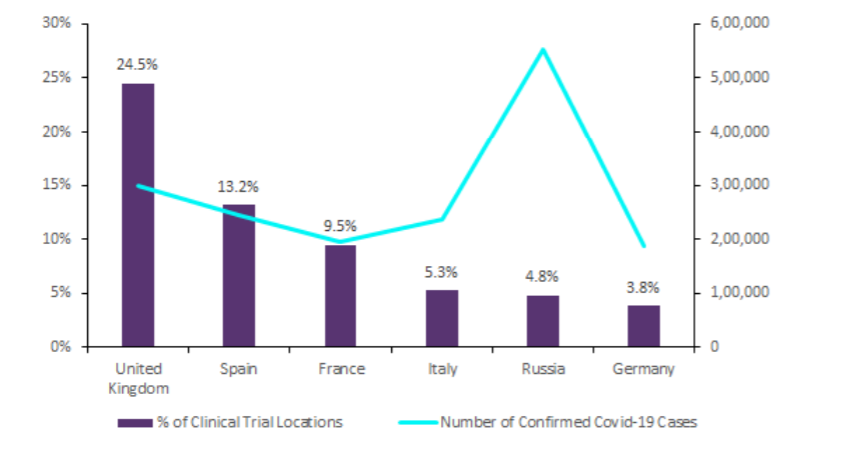
The Phase distribution of clinical trials suggests sponsors are focusing primarily on repurposing available therapies as opposed to working solely with investigational medicinal products (IMPs). Given the imminent threat to life posed by Covid-19, this is to be expected. Notably, the top six countries with the highest number of clinical trials also have the greatest number of Covid-19 cases in Europe, thereby highlighting that level of research directly correlates with the clinical need for effective therapies. As shown in Figure 1, the UK dominates the Covid-19 research space, accounting for the highest number of clinical trials (24.5%), followed by Spain (13.2%), France (9.5%), Italy (5.3%), Russia (4.8%), and Germany (3.8%).
Assistance Publique – Hopitaux de Paris is the top sponsor with 4.7% of European Covid-19 trials, more than double that of the University of Oxford, which sponsors the second largest number of trials (2.3%). Assistance Publique – Hopitaux de Paris utilises a range of therapy types in its studies, but it appears to have a focus on monoclonal antibodies as a treatment option for Covid-19.
Research efforts by the University of Oxford are mainly focused on finding a viable vaccine. The institute has co-partnered with AstraZeneca to develop the Covid-19 vaccine AZD1222. While efficacy results are yet to be published, a Phase III study is underway and production capacity has been up-scaled, suggesting there have been positive findings concerning safety and efficacy in the early phase trials.
Despite mixed data form the hydroxychloroquine studies and several studies being terminated, new trials using hydroxychloroquine with or without additional drugs continue to expand. It is the most widely used drug in Covid-19 trials worldwide and trials in Europe follow the same trend, with 14.4% of all European Covid-19 trials investigating the agent, more than double that of second-place tocilizumab (6.3%), as displayed by Figure 2. A Phase III study sponsored by the IHU-Mediterranee Infection in France had shown hydroxychloroquine to be effective in reducing the Covid-19 viral load, with its effect reinforced by azithromycin. This trial has gained significant media attention despite having several limitations in its trials, including sample size.
One of the largest Covid-19 clinical trials (RECOVERY) in Europe, which is sponsored by the University of Oxford, is investigating hydroxychloroquine as a therapy. However, the hydroxychloroquine arm halted enrolment after a review of the data showed that the treatment provided no viable benefits on mortality, disease course, or hospital stay. The RECOVERY trial had a vastly greater patient population, giving more credence to the results as compared with the earlier IHUMediterranee.
Recently, the Medicines and Healthcare Products Regulatory Agency (MHRA) has announced that it is instructing companies and organizations sponsoring UK clinical trials involving the use of hydroxychloroquine in Covid-19 to suspend the enrolment of patients. The MHRA decision stems from a review of the latest available data, including the RECOVERY trial, in which hydroxychloroquine provided no viable benefits in hospitalised patients. Furthermore, FDA has revoked hydroxychloroquine’s Emergency Use Authorization on the basis of current evidence which determined that the drug does not display adequate effectiveness in treating Covid-19, so the benefits of taking hydroxychloroquine do not outweigh the risks. It is therefore expected that a significant decline in the number of new trials investigating hydroxychloroquine as a Covid-19 therapy will occur. This is supported by the manner in which 71.4% of all Suspended, Terminated, or Withdrawn trials involve hydroxychloroquine.
Interestingly, researchers from the RECOVERY trial announced one of the other therapies being investigated, dexamethasone, has been shown to reduce the risk of death in patients on ventilators by one-third and reduce the risk of deaths on patents on oxygen by one-fifth. The new findings demonstrate the importance of the RECOVERY trial in identifying which treatments are or are not effective.
The significant amount of research effort across Europe signifies a shared goal in the search for effective therapies. Nevertheless, only 5.2% of trials have announced some form of efficacy results, which suggests the research is still in the early stages, and a continuation in a global effort is required to identify effective treatments to address the urgent need for effective therapies.