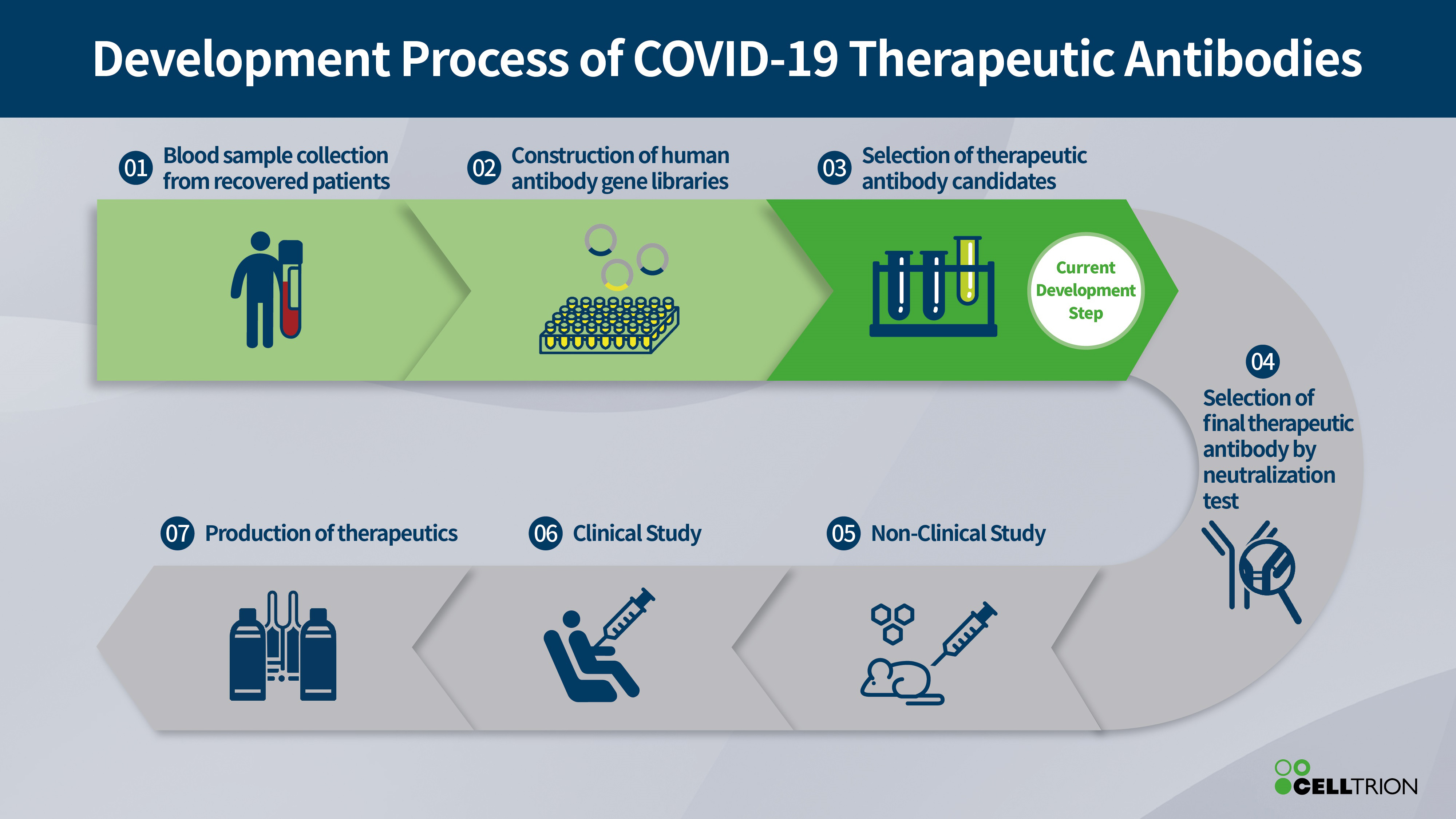
CAMBRIDGE, Mass. and TARRYTOWN, N.Y., March 16, 2020 /PRNewswire/ — Sanofi and Regeneron Pharmaceuticals, Inc. today announced they have started a clinical program evaluating Kevzara® (sarilumab) in patients hospitalized with severe COVID-19. Kevzara is a fully-human monoclonal antibody that inhibits the interleukin-6 (IL-6) pathway by binding and blocking the IL-6 receptor. IL-6 may play a role in driving the overactive inflammatory response in the lungs of patients who are severely or critically ill with COVID-19 infection. The role of IL-6 is supported by preliminary data from a single-arm study in China using another IL-6 receptor antibody.

This U.S.-based trial will begin at medical centers in New York, one of the epicenters of the U.S. COVID-19 outbreak, and will assess the safety and efficacy of adding Kevzara to usual supportive care, compared to supportive care plus placebo. The multi-center, double-blind, Phase 2/3 trial has an adaptive design with two parts and is anticipated to enroll up to 400 patients. The first part will recruit patients with severe COVID-19 infection across approximately 16 U.S. sites, and will evaluate the impact of Kevzara on fever and patients’ need for supplemental oxygen. The second, larger part of the trial will evaluate the improvement in longer-term outcomes including preventing death and reducing the need for mechanical ventilation, supplemental oxygen and/or hospitalization.
“At Sanofi, we are taking a leading role in addressing the global challenge of COVID-19 disease. We believe that there is scientific evidence to suggest that Kevzara may be a potentially important treatment option for some patients, and this trial will provide the well-controlled, rigorous scientific data we need to determine if IL-6 inhibition with Kevzara is better than current supportive care alone. Additionally, we expect to rapidly initiate trials outside the U.S. in the coming weeks, including areas most affected by the pandemic such as Italy,” said John Reed, M.D., Ph.D., Sanofi’s Global Head of Research and Development. “In addition to Kevzara, Sanofi Pasteur, the vaccines global business unit of Sanofi, is leveraging previous development work for a SARS vaccine as part of our goal to quickly develop a COVID-19 vaccine.”
Scientists have preliminary evidence that IL-6 may play a key role in driving the inflammatory immune response that causes acute respiratory distress syndrome (ARDS) in patients critically ill from COVID-19. Initial non-peer reviewed results from a single-arm, 21-patient Chinese trial found COVID-19 patients experienced rapidly reduced fevers and 75% of patients (15 out of 20) reduced their need for supplemental oxygen within days of receiving another IL-6 receptor antibody (tocilizumab). Based on these results, China recently updated its COVID-19 treatment guidelines and approved the use of that IL-6 inhibitor to treat patients with severe or critical disease.
“To initiate this trial quickly, Regeneron and Sanofi have worked closely with the U.S. Food and Drug Administration and the Biomedical Advanced Research and Development Authority, otherwise known as the FDA and BARDA ,” said George D. Yancopoulos, M.D., Ph.D., Co-founder, President and Chief Scientific Officer of Regeneron. “Data from China suggest that the IL-6 pathway may play an important role in the overactive inflammatory response in the lungs of patients with COVID-19. Despite this encouraging finding, it’s imperative to conduct a properly designed, randomized trial to understand the true impact. Our trial is the first controlled trial in the U.S. to evaluate the effect of IL-6 inhibition prospectively in COVID-19 patients. In addition to our Kevzara program, Regeneron is also rapidly advancing a novel antibody cocktail for the prevention and treatment of COVID-19, which we hope to have available for human testing this summer. Both of these programs are made possible by our unprecedented end-to-end antibody discovery, development and manufacturing technologies, starting with our proprietary VelocImmune human antibody mouse, and incorporating our associated rapid manufacturing technologies designed to select and produce the best neutralizing antibodies. Collectively, these technologies expedite a typically years-long process into a matter of months. This same technology was applied to the Ebola virus, where our therapy, REGN-EB3, was shown to dramatically improve survival in infected patients last year.”
In late 2019, Regeneron and Sanofi announced their intent to simplify the joint collaboration for Kevzara, which is expected to be finalized in the first-quarter of 2020. The companies will continue to collaborate on COVID-19 and other related ARDS development, with Regeneron leading U.S.-based work and Sanofi leading work outside of the U.S.
The use of Kevzara to treat the symptoms of COVID-19 is investigational and has not been fully evaluated by any regulatory authority.
About the Trial
This Phase 2/3, randomized, double-blind, placebo-controlled trial uses an adaptive design to evaluate the safety and efficacy of Kevzara in adults hospitalized with serious complications from COVID-19. To enter the trial, patients must be hospitalized with laboratory-confirmed COVID-19 that is classified as severe or critical, or who are suffering from multi-organ dysfunction. All patients must have pneumonia and fever. After receiving the study dose, patients will be assessed for 60 days, or until hospital discharge or death.
In the Phase 2 part of the trial, patients will be randomized 2:2:1 into three groups: Kevzara high dose, Kevzara low dose and placebo. The primary endpoint is reduction of fever and the secondary endpoint is decreased need for supplemental oxygen.
The Phase 2 findings will be utilized in an adaptive manner to determine transition into Phase 3, helping to determine the endpoints, patient numbers and doses. The second, larger part of the trial will evaluate the improvement in longer-term outcomes including preventing death and reducing the need for mechanical ventilation, supplemental oxygen and/or hospitalization.
If the trial continues with all three treatment arms to the end, it is expected to enroll approximately 400 patients, depending on the status of the COVID-19 outbreak and the proportion of patients with severe COVID-19 and high levels of IL-6.
About Kevzara® (sarilumab) Injection
Kevzara was jointly developed by Sanofi and Regeneron under a global collaboration agreement. Kevzara is a fully-human monoclonal antibody. Kevzara binds specifically to the IL-6 receptor, and has been shown to inhibit IL-6-mediated signaling. IL-6 is a signaling protein produced in increased quantities in patients with rheumatoid arthritis and has been associated with disease activity, joint destruction and other systemic problems. It is also being investigated for its ability to reduce the overactive inflammatory immune response associated with COVID-19.