Progress on new pump hardware, a wider age indication for its new insulin dosing algorithm, and the ability to control dosing from a phone app are among Tandem Diabetes’ focuses for 2020, execs outlined on an earnings call Monday.
Annual revenue nearly doubled to about $362 million in 2019 from a year earlier, Tandem reported after markets closed, well exceeding the $270 million high predicted early last year. Over the course of those 12 months, the company shipped more than 73,000 of its pumps, up 113% compared to 2018.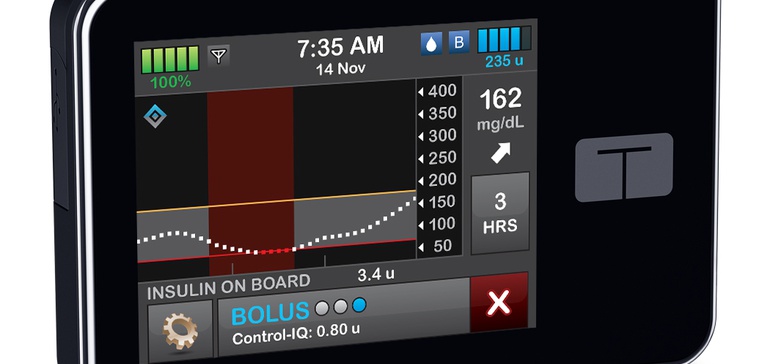
Still, fourth quarter results — which marked comparatively underwhelming 42% year-over-year sales growth — didn’t blow Wall Street analysts away, coming in largely as expected. Shares were down 7% in late morning trade Tuesday.
Stifel analysts noted further share price appreciation is contingent nearly entirely on better-than-expected revenue.
“Multiple variables all need to skew in [Tandem’s] favor, which while possible, doesn’t yet feel probable/inevitable to us,” they wrote.
Those factors include a high level of new pump customers, more transitions from patients currently on multiple daily injection therapy, and acceleration of competitor switches.
Execs said the company now has close to 20% of the U.S. insulin pump market and predict sales to reach up to $465 million in 2020, forecasting growth between 24% and 28%. Gross margins rose five points over 2019 to 54%, where Tandem expects them to stay steady in 2020.
Ability to administer bolus doses of insulin via a phone is “the number one feature that people would desire,” CEO John Sheridan told analysts on an investor call. An FDA filing is planned for this summer for a new mobile app that allows for remote bolusing.
Sheridan said the submission will be “a bit of uncharted territory as there is not a commercial precedent for mobile control from an insulin pump using an unrestricted mobile device,” making FDA timing hard to predict, though it estimates the feature will be available in the second half of this year.
Tandem also noted its future FDA submission of a smaller insulin pump called t:sport will come in two filings, because pump users will have the choice to either operate the device via a phone or from a separate handheld device. The company plans to prioritize the latter and file it this summer, with plans to submit the full control from a mobile app version later. Tandem does not plan to launch the new pump until each of the two submissions are OK’ed by FDA and is currently targeting the first half of 2021.
Impact of Control-IQ
The company previously cautioned investors that some customers may have been waiting until FDA greenlighted its automated insulin dosing algorithm known as Control-IQ to buy a new pump.
“It is difficult to quantify the impact of the timing of Control-IQ had on 2019,” CFO Leigh Vosseller said Monday, but said the company estimates around 2,000 customers will purchase a pump in 2020 “who would have otherwise purchased a pump last year.”
After a review that lasted just shy of five months, FDA in December authorized Control-IQ through the De Novo pathway — designed for medical technologies that can be sufficiently regulated as Class I or II devices with general and/or special controls, but for which no predicate device exists.
The go-ahead for Control-IQ created the third and final category necessary for having an automated insulin dosing system, following FDA’s designation of Dexcom’s G6 CGM as an integrated continuous glucose monitor and of Tandem’s own t:slim X2 insulin pump as an alternate controller enabled pump.
The company plans to file a regulatory submission this quarter to allow Control-IQ for people as young as age 6. When first awarded the De Novo, Tandem’s technology was authorized for use in individuals 14 years old and up.
Tandem also plans to roll out improvements to the Control-IQ algorithm in 2021, which it’s having to work through with regulators. “We are working with the FDA to segment various features by level of clinical risks to ultimately determine the regulatory path that will allow us to make incremental updates to our Control-IQ technology and then deliver new benefits to our customers on a regular basis,” Sheridan said.
If competitor Insulet’s intentions to bring its own automated insulin delivery system to market in 2020 go according to plan, Tandem will likely face stiffer competition at the end of the year. Major continuous glucose monitor manufacturers Dexcom and Abbott each outlined plans last week to integrate their devices with Insulet’s system, called Horizon. Insulet is scheduled to report earnings after markets close Tuesday.
Control-IQ currently operates in tandem with Dexcom’s G6 CGM. Tandem and Abbott announced last October they “intend to develop and commercialize integrated diabetes solutions” but offered few details on when an Abbott-Tandem-compatible system may be available and what it might look like. On Monday’s call, Sheridan offered few additional details, saying the companies are still working on their agreement and he’s confident “we’re going to get this thing been taken care of in the near future.”
While the pharmacy has been an increasingly popular point of sale for CGM counterparts, Vosseller told analysts Tandem isn’t necessarily looking to move away from its durable medical equipment selling model.