Dive Brief:
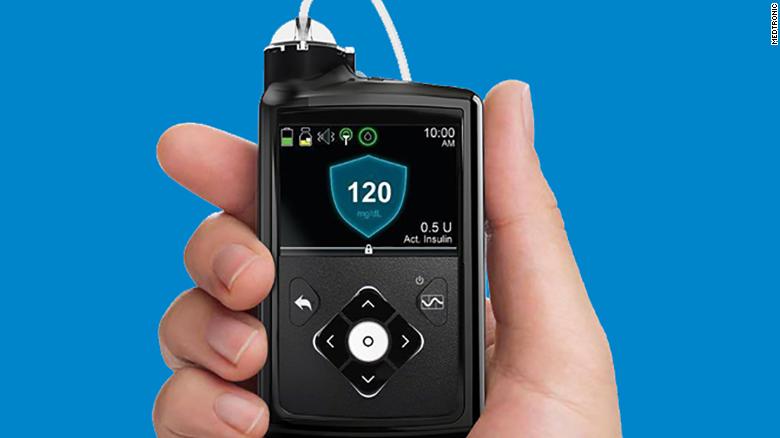
- A Medtronic insulin pump hardware malfunction that prompted the company to alert certain MiniMed customers late last year has been linked to 26,421 complaints, 2,175 injuries and one death, FDA said in a noticeissued Wednesday.
- Medtronic warned customers Nov. 21 that a potential missing or broken retainer ring, which ordinarily helps secure the insulin cartridge’s position in the pump’s reservoir compartment, could lead to under or over delivery of insulin, thereby increasing risk for hypoglycemia and hyperglycemia and ultimately loss of consciousness, seizure or death.
- Now the agency has categorized the action affecting more than 320,000 U.S. devices as a Class I recall, reflecting the concern that use of affected devices may result in serious injury or death.
Dive Insight:
In total, 322,005 devices in the U.S. are covered by the recall, FDA detailed Wednesday. Affected devices include all 630G pumps distributed between September 2016 and October 2019, and 670G pumps distributed between June 2017 and August 2019.
The latter model was Medtronic’s big diabetes innovation approved by FDA in 2016 as the first automated insulin delivery device. In the years since, companies like Tandem and Insulet have broken through with their own diabetes management products, but Medtronic hopes to retake share with its forthcoming 780G pump.
That device, which Medtronic is calling a personalized hybrid closed loop system, is designed to automatically deliver correction bolus doses in anticipation of, or when a user experiences, prolonged high glucose levels. Medtronic expects to present pivotal trial data on the device at the American Diabetes Association meeting in June.
There are no new instructions to customers, a Medtronic spokesperson confirmed Wednesday. The company maintained earlier guidance that customers should contact Medtronic for a replacement pump if, upon examination, the reservoir doesn’t lock into the pump or if the retainer ring is loose, damaged or missing. Medtronic also advised that users check for proper function of the retainer ring if the pump is dropped by accident and at every set change.
In the past week, FDA also sent a Class I recall notice regarding nearly 3,600 GE Healthcare anesthesia systems that may have problems with mechanical ventilation.
Medtronic is scheduled to report recent financial results and discuss plans for the coming months during an earnings call Tuesday.