For 20 years, the U.S. government has urged companies, universities, and other institutions that conduct clinical trials to record their results in a federal database, so doctors and patients can see whether new treatments are safe and effective. Few trial sponsors have consistently done so, even after a 2007 law made posting mandatory for many trials registered in the database. In 2017, the National Institutes of Health (NIH) and the Food and Drug Administration (FDA) tried again, enacting a long-awaited “final rule” to clarify the law’s expectations and penalties for failing to disclose trial results. The rule took full effect 2 years ago, on 18 January 2018, giving trial sponsors ample time to comply. But a Science investigation shows that many still ignore the requirement, while federal officials do little or nothing to enforce the law.
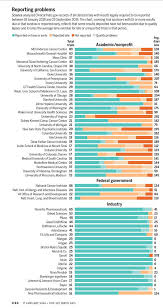
Science examined more than 4700 trials whose results should have been posted on the NIH website ClinicalTrials.gov under the 2017 rule. Reporting rates by most large pharmaceutical companies and some universities have improved sharply, but performance by many other trial sponsors—including, ironically, NIH itself—was lackluster. Those sponsors, typically either the institution conducting a trial or its funder, must deposit results and other data within 1 year of completing a trial. But of 184 sponsor organizations with at least five trials due as of 25 September 2019, 30 companies, universities, or medical centers never met a single deadline. As of that date, those habitual violators had failed to report any results for 67% of their trials and averaged 268 days late for those and all trials that missed their deadlines. They included such eminent institutions as the Harvard University–affiliated Boston Children’s Hospital, the University of Minnesota, and Baylor College of Medicine—all among the top 50 recipients of NIH grants in 2019.
The violations cover trials in virtually all fields of medicine, and the missing or late results offer potentially vital information for the most desperate patients. For example, in one long-overdue trial, researchers compared the efficacy of different chemotherapy regimens in 200 patients with advanced lymphoma; another—nearly 2 years late—tests immunotherapy against conventional chemotherapy in about 600 people with late-stage lung cancer.
Other leading NIH grantees did only slightly better in Science’s analysis based on data collected from the TrialsTracker website of the University of Oxford, which automatically mines information from ClinicalTrials.gov. The University of Texas MD Anderson Cancer Center and the Mayo Clinic both failed to report results on time, or at all, in about two-thirds of their trials. Yale University failed to do so in 84% of its trials. NIH’s own institutes also had a bad record. They are directly responsible for reporting results when they sponsor studies done by agency staff or some grantees, and the top four NIH institute sponsors, taken together, reported results late or not at all in more than six of every 10 trials Science looked at.
Contacted for comment, none of the institutions disputed the findings of this investigation. In all 4768 trials Science checked, sponsors violated the reporting law more than 55% of the time. And in hundreds of cases where the sponsors got credit for reporting trial results, they have yet to be publicly posted because of quality lapses flagged by ClinicalTrials.gov staff (see sidebar).
Although the 2017 rule, and officials’ statements at the time, promised aggressive enforcement and stiff penalties, neither NIH nor FDA has cracked down. FDA now says it won’t brandish its big stick—penalties of up to $12,103 a day for failing to report a trial’s results—until after the agency issues further “guidance” on how it will exercise that power. It has not set a date. NIH said at a 2016 briefing on the final rule that it would cut off grants to those who ignore the trial reporting requirements, as authorized in the 2007 law, but so far has not done so.
Many scientists who conduct clinical trials, and their sponsors or funders, have downplayed concerns about late or missing results in ClinicalTrials.gov. Researchers, doctors, and patients can instead learn about trial outcomes from peer-reviewed publications, they say. But thousands of trials are never published, particularly when they find treatments ineffective, history has shown. ClinicalTrials.gov also uses a common format, allowing relatively easy comparisons of results across trials that journal articles rarely make possible. Doctors, researchers, and potential trial participants rely on the site, to judge from its 215 million monthly page views.
Deborah Zarin, a physician at Brigham and Women’s Hospital and Harvard who headed ClinicalTrials.gov between 2005 and 2018, says the Science findings show failures of the research culture, FDA, and NIH. “If this was a priority for the leadership of NIH, then they could ensure that high-quality, timely reporting happened all of the time,” says Zarin, an NIH-paid research consultant for the database. “You can set up processes so trial reporting is an expectation. You can’t pass ‘go’ and collect $200 until this is done.”
Zarin, who works in a program to advance clinical research, adds that the problem persists because “reporting to ClinicalTrials.gov is frequently seen by sponsors, funders, and trialists as an annoying administrative and perhaps legal burden, not a scientific imperative. Human nature being what it is, people follow the requirements when forced to do so.”
NIH and FDA officials do not seem inclined to apply that pressure. Lyric Jorgenson, NIH deputy director for science policy, says her agency has been “trying to change the culture of how clinical trial results are reported and disseminated; not so much on the ‘aha, we caught you,’ as much as getting people to understand the value, and making it as easy as possible to share and disseminate results.” To that end, she says, ClinicalTrials.gov staff have educated researchers about the website and improved its usability.
As for FDA, Patrick McNeilly, an official at the agency who handles trial enforcement matters, recently told an industry conference session on ClinicalTrials.gov that “FDA has limited resources, and we encourage voluntary compliance.” He said the agency also reviews reporting of information on ClinicalTrials.gov as part of inspections of trial sites, or when it receives complaints.
McNeilly declined an interview request, but at the conference he discounted violations of ClinicalTrials.gov reporting requirements found by journalists and watchdog groups. “We’re not going to blanketly accept an entire list of trials that people say are noncompliant,” he said. Such determinations require “nonpublic information” submitted to the agency by trial sponsors. In response to Science’s findings, a spokesperson said an absence of posted results on ClinicalTrials.gov did not mean a trial sponsor has broken the 2007 law.
Yet that law and the 2017 final rule detail only a few exemptions that would allow trial sponsors to withhold results on the basis of nonpublic information. The very few registered trials that qualify for those exemptions are not flagged as violators by TrialsTracker or in Science’s analysis.
CONGRESS APPROVED THE CREATION OF ClinicalTrials.gov in 1997, after allegations that patients were harmed because companies withheld evidence showing their medicines were ineffective or hazardous. A widely cited case involved the GlaxoSmithKline antidepressant Paxil (paroxetine). According to legal filings and a report in The BMJ, the firm held secret data showing that in clinical trials the drug was ineffective and caused suicidal thoughts in teenagers, yet encouraged doctors to prescribe it for young people.
Registration was only required initially for trials of treatments for serious or life-threatening diseases. But the 2007 law, the Food and Drug Administration Amendments Act, required sponsors to register a much broader range of trials within 21 days of enrolling the first patient, and to post summary results, adverse events, and other data to ClinicalTrials.gov within 1 year of collecting the last patient data. Although many trials, such as industry-sponsored early-stage evaluations of drug safety, are exempt from reporting, about 326,000 have been registered, and results have been posted for more than 40,000.
Yet until 2015, even the most active investigators at clinical research institutions treated the law more as a suggestion—not surprising given that the government enforced no penalties and did not publicly identify violators. A report on the news website STAT by this author and Talia Bronshtein first drew significant attention to specific trial sponsors—companies, government agencies, universities, and individuals—that routinely ignored reporting requirements. It sparked immediate improvement, according to NIH. (Those same authors documented some of that improvement in a 2018 STAT article.)
At a 2016 press briefing, NIH and FDA rolled out the final rule, aimed at boosting even greater compliance with the 2007 law. It took effect in January 2017, with first deadlines for results, and ostensibly enforcement, 1 year later. Then–FDA Commissioner Robert Califf said it would thereafter “be pretty hard to hide that you are doing a clinical trial or hide the result.” FDA, he vowed, was finally prepared, if necessary, to enforce the daily $10,000 penalty for noncompliance allowed under the law. (Adjusted for inflation, that figure recently rose above $12,000.)
“I don’t think anybody wants to be on the wall of shame,” NIH Director Francis Collins said at the press event, promising that NIH would publicly flag reporting violations on ClinicalTrials.gov itself.
“We are serious about this,” Collins said, threatening for the first time to enforce provisions of the 2007 law that allow NIH to rescind funding to grantees who violate the statute. “It’s hard to herd cats, but you can … take their food away,” he said. “This is about maintaining the trust that we have with participants in clinical trials. … If we fail to live up to that expectation, then that is an ethical failure.”
Three years later, TrialsTracker conservatively estimates that FDA could have collected more than $6 billion in ClinicalTrials.gov penalties so far. The agency has yet to demand a single dollar. And despite more than 2600 trials for which results are overdue or were filed late, NIH has yet to withhold a single grant as a result or post a single violation notice on ClinicalTrials.gov. No “wall of shame” exists.
“Public-facing websites run by the government should be accurate. That’s not asking much,” Senator Chuck Grassley (R–IA), who advocated for the 2007 law, wrote in an email after reviewing a summary of the Science findings. “It’s a question of basic management and agency competence. The government has a duty to police its work product, especially because the public trusts .gov websites will be accurate and reliable.”
To physician Ben Goldacre, who directs the Oxford program behind TrialsTracker, “The lack of urgency is really troubling.”